
Genome-wide relationship between histone H3 lysine 4 mono- and tri-methylation and transcription factor binding
- A. Gordon Robertson1,4,
- Mikhail Bilenky1,4,
- Angela Tam1,
- Yongjun Zhao1,
- Thomas Zeng1,
- Nina Thiessen1,
- Timothee Cezard1,
- Anthony P. Fejes1,
- Elizabeth D. Wederell2,
- Rebecca Cullum2,
- Ghia Euskirchen3,
- Martin Krzywinski1,
- Inanc Birol1,
- Michael Snyder3,
- Pamela A. Hoodless2,
- Martin Hirst1,
- Marco A. Marra1, and
- Steven J.M. Jones1,5
- 1 BC Cancer Agency Genome Sciences Centre, Vancouver V5Z 4S6, Canada;
- 2 Terry Fox Laboratory, BC Cancer Agency, Vancouver V5Z 1L3, Canada;
- 3 Department of Molecular, Cellular and Developmental Biology, Yale University, New Haven, Connecticut 06520, USA
-
↵4 These authors contributed equally to this work.
Abstract
We characterized the relationship of H3K4me1 and H3K4me3 at distal and proximal regulatory elements by comparing ChIP-seq profiles for these histone modifications and for two functionally different transcription factors: STAT1 in the immortalized HeLa S3 cell line, with and without interferon-gamma (IFNG) stimulation; and FOXA2 in mouse adult liver tissue. In unstimulated and stimulated HeLa cells, respectively, we determined ∼270,000 and ∼301,000 H3K4me1-enriched regions, and ∼54,500 and ∼76,100 H3K4me3-enriched regions. In mouse adult liver, we determined ∼227,000 and ∼34,800 H3K4me1 and H3K4me3 regions. Seventy-five percent of the ∼70,300 STAT1 binding sites in stimulated HeLa cells and 87% of the ∼11,000 FOXA2 sites in mouse liver were distal to known gene TSS; in both cell types, ∼83% of these distal sites were associated with at least one of the two histone modifications, and H3K4me1 was associated with over 96% of marked distal sites. After filtering against predicted transcription start sites, 50% of ∼26,800 marked distal IFNG-stimulated STAT1 binding sites, but 95% of ∼5800 marked distal FOXA2 sites, were associated with H3K4me1 only. Results for HeLa cells generated additional insights into transcriptional regulation involving STAT1. STAT1 binding was associated with 25% of all H3K4me1 regions in stimulated HeLa cells, suggesting that a single transcription factor can interact with an unexpectedly large fraction of regulatory regions. Strikingly, for a large majority of the locations of stimulated STAT1 binding, the dominant H3K4me1/me3 combinations were established before activation, suggesting mechanisms independent of IFNG stimulation and high-affinity STAT1 binding.
Enhancers are regulatory elements that activate transcription by interacting with target promoters, typically by acting over large genomic distances (Bondarenko et al. 2003; Arnosti and Kulkarni 2005; Szutorisz et al. 2005). Locations of putative enhancers have been inferred by distal binding sites for the cofactor EP300 (also known as p300) (Hatzis and Talianidis 2002 Kalkhoven 2004; Heintzman et al. 2007), and by filtering sets of distal DNase I hypersensitive sites (DHS) to remove potential promoters and insulators (Barski et al. 2007; Boyle et al. 2008; Wang et al. 2008). Five recent studies, which used different platforms and different enhancer surrogates, appear to give divergent results for whether histone modifications can distinguish active promoters from distal enhancers. Initially, data from chromatin immunoprecipitation with microarray hybridization (ChIP-chip) for the 1% of the human genome represented by the ENCODE regions indicated that the monomethylated histone H3 lysine 4 (H3K4me1), but not the trimethylated form (H3K4me3), was enriched around distal EP300 sites, while both modifications were enriched at promoters (The ENCODE Project Consortium 2007; Heintzman et al. 2007). However, data from chromatin immunoprecipitation with massively parallel sequencing (ChIP-seq) for human T-cells subsequently showed that both H3K4me1 and H3K4me3, along with certain other modifications and the histone variant H2A.Z, were enriched at putative enhancers that were inferred from filtered distal DHS (Barski et al. 2007; Wang et al. 2008). Finally, H3K4me1 was enriched relative to H3K4me3 in ChIP-seq data, at putative enhancers that were inferred by filtering a more comprehensive set of T-cell DHS (Boyle et al. 2008). Together, these studies indicated that different types of distal elements are associated with different combinations of modifications, but left unresolved whether H3K4me1 and H3K4me3 can be used to distinguish distal enhancers from active promoters.
ChIP-seq profiles for protein–DNA association can identify distal and proximal regulatory elements with high spatial resolution. In the work described here, we took advantage of this resolution to assess, for the first time, the direct relationship of H3K4me1 and H3K4me3 at regulatory elements inferred from profiles for two functionally different transcription factors. We assessed STAT1 in the immortalized HeLa S3 cell line, with and without interferon-gamma (IFNG) stimulation (Robertson et al. 2007), and FOXA2 in a normal, non-immortalized tissue, mouse adult liver (Wederell et al. 2008). We profiled H3K4me1 and H3K4me3 in these cells and determined combinations of modifications that were associated with transcription factor binding. We compared our results to those noted above for putative enhancers inferred by EP300 sites and by filtered DHS. Our results indicate that H3K4me1 is a dominant mark for active and potentially active distal regulatory regions; at the same time, a subset of distal regions is associated with both H3K4me1 and H3K4me3, and the relative size of this subset may vary between different biological systems.
Results
Histone modification profiles were saturated
We sequenced chromatin immunoprecipitated DNA and identified regions that were enriched in STAT1 binding and in H3K4me1 and H3K4me3 in unstimulated and IFNG-stimulated HeLa S3 cells, and in mouse adult liver (Table 1). The relationship between sequencing depth and the number of modified histone regions, at a constant false discovery rate (FDR) of ∼0.01, indicated that we had generated saturated profiles, that is, profiles that determined essentially all enriched regions that were available under the experimental conditions (Supplemental Fig. SA1A,B). In contrast, we observed nonsaturating behavior for the transcription factors STAT1 and FOXA2 (Supplemental Fig. SA1C,D).
Summary data for ChIP-seq regions enriched in STAT1 or FOXA2, H3K4me1, or H3K4me3
H3K4me1 was the dominant modification for distal STAT1 sites, EP300 sites, and predicted enhancers in HeLa cells
Using a 1-kb distance threshold (Supplemental Fig. GA1A), we found that 32.7% of the overall ∼70,300 STAT1 binding sites in stimulated HeLa cells were associated with H3K4me1, 47.1% were associated with both H3K4me1 and H3K4me3, 6.9% were associated with H3K4me3 but not H3K4me1, and 13.4% were associated with neither modification (Figs. 1E, 2C).
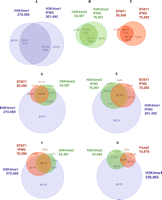
Association and concordance rates. Concordance rates between unstimulated and IFNG-stimulated HeLa cells for H3K4me1 (A), H3K4me3 (B), and STAT1 (C), using FDR ∼0.01 profiles, with random expectations in parentheses. Association rates between STAT1 and H3K4me1/me3 in unstimulated (D) and stimulated (E) HeLa cells. (F) Association rates between STAT1 in IFNG-stimulated cells and H3K4me1/H3K4me3 in unstimulated cells. (G) Association rates between FOXA2, H3K4me1, and H3K4me3 in adult mouse liver. (A–C) Concordance rates relative to numbers of both unstimulated and stimulated regions (e.g., for H3K4me1, 72.0% and 67.4%). (D–G) Percentages in red are relative to the TF (e.g., in E, 32.7% of IFNG-stimulated STAT1 binding sites are associated with H3K4me1 and not H3K4me3), while those in blue and green are relative to H3K4me1 and H3K4me3, respectively, and refer to modified regions that are not associated with the TF (e.g., in E, 7.1% of the H3K4me1 regions that are not associated with STAT1 are associated with H3K4me3).
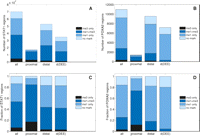
Relationships between regions enriched in STAT1 or FOXA2 and in H3K4me1 and H3K4me3. (A,C) Number and fraction of all, proximal, and distal STAT1 binding sites, and distal binding sites filtered by predicted TSS (DEE) in IFNG-stimulated HeLa cells. (B,D) As for STAT1, but for distal FOXA2 binding sites in mouse adult liver. A binding site was distal if the location of its maximum coverage was farther than ±2.5 kb from a transcriptional start site of UCSC known gene.
We then assessed whether the high positive spatial correlation between STAT1 sites and H3K4me1 and H3K4me3 regions in stimulated cells was due to these sites being closely associated with promoters. A distance threshold of ±2.5 kb (The ENCODE Project Consortium 2007; Heintzman et al. 2007) relative to transcription start sites (TSS) for UCSC hg18 known genes (Hsu et al. 2006) identified 75.4% of the overall 70,300 STAT1 sites as distal and 24.6% as proximal. Of the ∼53,000 distal STAT1 sites, 39.9% were associated with H3K4me1 but not H3K4me3, 40.5% were associated with both H3K4me1 and H3K4me3, 3.2% were associated with H3K4me3 but not H3K4me1, and 16.6% were associated with neither modification (Fig. 2A,C). Of the ∼17,300 proximal STAT1 sites, 11.6% were associated with H3K4me1 but not H3K4me3, 67.3% were associated with both H3K4me1 and H3K4me3, 17.2% were associated with H3K4me3 but not H3K4me1, and 3.8% were associated with neither modification.
We tested whether our ChIP-seq profiles were consistent with the combinations of H3K4me1 and H3K4me3 that had been reported for distal EP300 sites and distal predicted enhancers from ChIP-chip data in ENCODE regions for HeLa cells (Heintzman et al. 2007). Again using a distance threshold of 1 kb (Supplemental Fig. EN1), we found that 37.2% of the 153 distal EP300 sites reported in that earlier work were associated with H3K4me1 but not H3K4me3, 46.4% were associated with both H3K4me1 and H3K4me3, 3.3% were associated with H3K4me3 but not H3K4me1, and 13.1% were associated with neither modification (Supplemental Fig. EN2). For the 284 distal predicted enhancer locations reported in that earlier work, 44.4% were associated with H3K4me1 but not H3K4me3, 34.2% were associated with both H3K4me1 and H3K4me3, 3.2% were associated with H3K4me3 but not H3K4me1, and 18.3% were associated with neither modification.
We can summarize these results as follows. Approximately 80% of ∼53,000 distal STAT1 sites in stimulated cells were associated with H3K4me1. Close to half of these H3K4me1-associated regions were associated with H3K4me1 but not H3K4me3, while the other half was associated with both. Proximal STAT1 sites were largely associated with both modifications. Results for distal STAT1 sites were similar to those determined from our profiles at the distal EP300 sites and distal predicted enhancers reported in the 1% of the human genome represented by ENCODE regions.
H3K4me1 was the dominant histone modification for distal FOXA2 sites in mouse adult liver
We used data for FOXA2 binding in mouse adult liver (Wederell et al. 2008) to assess whether the histone modification patterns that were associated with STAT1 in HeLa cells were unique to that transcription factor (TF) and cell line or were more general (Table 1).
For individual histone modifications, 81.0% (random expectation 13.5%) of the ∼11,000 FOXA2 binding sites were within 1 kb (Supplemental Fig. GA1B) of H3K4me1 regions, and 25.5% (random expectation 2%) were associated with H3K4me3 regions (Fig. 1G). When we assessed combinations of modifications, we found that 58.7% of the FOXA2 sites were associated with H3K4me1 but not H3K4me3, 22.3% were associated with both H3K4me1 and H3K4me3, 3.2% were associated with H3K4me3 but not H3K4me1, and 15.8% were associated with neither modification (Fig. 2B,D).
We again used a distance of ±2.5 kb relative to TSS of UCSC known genes to distinguish ∼9600 (87.2%) distal and ∼1400 (12.8%) proximal FOXA2 sites, and compared combinations of histone modifications that were associated with the two groups (Fig. 2B,D). Of distal sites, 64.1% were associated with H3K4me1 but not H3K4me3, 16.4% were associated with both H3K4me1 and H3K4me3, 1.9% were associated with H3K4me3 but not H3K4me1, and 17.5% were associated with neither modification. For proximal sites, 11.8% were associated with H3K4me1 but not H3K4me3, 62.2% were associated with both H3K4me1 and H3K4me3, 22.1% were associated with H3K4me3 but not H3K4me1, and 4.0% had neither modification.
We can summarize the association results for STAT1 in HeLa cells and FOXA2 in mouse adult liver by noting that ∼80% of distal binding sites were associated with the same two dominant H3K4me1–me3 patterns. However, a larger fraction of distal FOXA2 sites was associated with H3K4me1 only (64% vs. 40%), and a smaller fraction was associated with both H3K4me1 and H3K4me3 (16% vs. 40%).
Filtering distal sites with predicted TSS and CAGE tags can change ratios of associated modifications
In previous work involving ChIP-seq profiles, UCSC known genes were considered to be an incomplete set of promoters, and input sets of DHS were filtered against RNA polymerase II sites or expression evidence that was distal to known gene TSS (Barski et al. 2007; Boyle et al. 2008; Wang et al. 2008). To support comparing our results to this work, we tested whether the histone modifications associated with our distal IFNG-stimulated STAT1 and FOXA2 binding sites were robust to similar filtering.
For the ∼53,000 IFNG-stimulated STAT1 binding sites that were distal to known gene TSS, we reclassified 33.9% as proximal to TSS of UCSC hg18 AceView, GENSCAN, and SwitchGear TSS predictions. Extending the filtering to include 123,000 CAGE tag clusters (Abeel et al. 2008) reclassified only an additional 1.5% of the ∼53,000 sites. Unexpectedly, given the rationale for DHS filtering, ratios of modifications for the reclassified sites were similar to ratios for distal sites, rather than to proximal sites (Fig. 2A,C; Supplemental Tables PT1, PT2; Supplemental Fig. PT1). Given this, the dominant combinations of modifications for distal STAT1 sites were insensitive to this filtering.
For the ∼9600 distal FOXA2 sites that were distal to known gene TSS, we reclassified 24.9% as proximal to TSS of UCSC mm8 SIB gene and GENSCAN predictions (Fig. 2B,D; Supplemental Tables PT3, PT4; Supplemental Fig. PT2). In contrast to results for STAT1 (IFNG), the reclassified group had a higher proportion of H3K4me3, consistent with expectations from DHS filtering. For the ∼7200 sites that remained distal, 75.9% were associated with H3K4me1 only, and only 4.1% with H3K4me1 and H3K4me3.
We noted that, for histone modification profiles thresholded with an FDR of ∼0.01, ∼17% of distal STAT1 and FOXA2 binding sites were associated with neither H3K4me1 nor H3K4me3 (Fig. 2C,D). Differences between the two cell types studied were highlighted when we considered TSS-filtered results for the ∼83% of distal sites that were associated with at least one mark. For these binding sites, ∼51% of filtered, distal IFNG-stimulated STAT1 sites, but >99% of the filtered distal FOXA2 sites, were marked by H3K4me1 only (Supplemental Tables PT2, PT4).
More stringent modification FDRs confirmed H3K4me1 as dominant at distal TF binding sites
Results from filtering distal STAT1 sites against predicted TSS encouraged us to assess whether the ratios of distal modification patterns were robust to thresholding modification profiles with more stringent FDR values.
Both distal STAT1 and FOXA2 binding sites were associated with less significant H3K4me3 regions and more significant H3K4me1 regions (Supplemental Figs. FD1, FD3). For distal STAT1 sites in stimulated HeLa cells, decreasing the FDR thresholds from ∼10−2 to ∼3 × 10−4 increased the fraction of STAT1 sites associated with H3K4me1 and not H3K4me3 from 39.7% to 46.1%, while decreasing the fraction associated with both H3K4me1 and H3K4me3 from 40.5% to 26.0% (Supplemental Tables FD1–FD3; Supplemental Fig. FD2). For distal FOXA2 sites, decreasing profile-specific FDR thresholds from ∼10−2 to ∼10−4 increased the fraction of FOXA2 sites that were associated with H3K4me1 and not H3K4me3 from 64.1% to 67.6%, while decreasing the fraction of sites that were associated with both H3K4me1 and H3K4me3 from 16.4% to 8.7% (Supplemental Tables FD4–FD6; Supplemental Fig. FD5).
These results suggested that distal TF binding sites that were marked by both modifications were associated with more H3K4me1 than H3K4me3, consistent with ChIP-seq results for filtered DHS (Barski et al. 2007; Wang et al. 2008).
For distal TF sites, we can compare results for STAT1 in stimulated cells and FOXA2 by considering the ratio of the number of distal sites that were associated with H3K4me1 to those associated with H3K4me3. As we decreased the FDR threshold from ∼10−2 to ∼10−4, the ratio increased from 1.8 to 2.5 for distal STAT1, but from 4.4 to 7.4 for distal FOXA2. Filtering against additional predicted TSS (above) identified ∼35,000 distal STAT1 sites that had an H3K4me1:H3K4me3 ratio of 1.9, but ∼7200 distal FOXA2 sites that had a ratio, 18.2, that was 10 times higher.
Missing modifications were likely due to factors other than read mappability
Even though our histone modification profiles appeared to be saturated, 16.6% of distal STAT1 sites in stimulated HeLa cells and 17.5% of distal FOXA2 sites in mouse liver were associated with neither H3K4me1 nor H3K4me3 at the default FDR threshold of ∼0.01 (Fig. 2C,D). We could resolve between a quarter and a half of the missing modifications by using a larger association distance than 1 kb (Supplemental Fig. GA1).
However, we generated ChIP-seq profiles using sequence reads that had been aligned to unique genomic locations, and reads cannot be aligned or mapped uniquely into certain repetitive genomic regions. Given this, we profiled the average read mappability around subsets of distal STAT1 sites in stimulated HeLa cells and FOXA2 sites in mouse adult liver to assess whether missing modifications were likely to be false negatives caused by local mappability deficits that reduced aligned read densities (Supplemental Fig. MP1). For both TFs, the mappability profiles were consistent with some asymmetric flanking H3K4me1 cases being caused by low flanking mappability. However, both STAT1 and FOXA2 sites had relatively high average flanking read mappabilities. This suggests that most cases in which a distal TF site lacked associated H3K4me1 were caused by the TF being associated either with no modifications, or with different modifications from those we profiled, rather than being false negatives caused by read mappability.
Distal regions flanked by H3K4me1 were enriched in known transcription factor binding site sequences
We noted that TF-associated H3K4me1 regions often occurred as a symmetric flanking pair (Figs. 3, 4A,C). Approximately 75% of ∼301,000 H3K4me1-enriched regions in stimulated HeLa cells and 94% of ∼227,000 regions in mouse liver were not associated with STAT1 and FOXA2, respectively (Fig. 1E,G). To demonstrate that distal H3K4me1-associated regions had sequence properties that were consistent with those of regulatory regions, we assessed whether a subset of genomic regions that were symmetrically flanked by this modification were enriched in known functional binding site sequences for mammalian transcription factors.
We first determined that H3K4me1-enriched regions that flanked STAT1 and FOXA2 locations were typically separated by a distance of between 200 and 1000 bp (Supplemental Figs. TF1, TF2). We used this distance range as a filter to identify ∼90,100 flanked regions in stimulated HeLa cells, ∼77,900 of which were distal, and ∼63,700 flanked regions in mouse adult liver, ∼54,100 of which were distal (Supplemental Table TF2). We then searched the genomic sequences within these regions for exact matches to known binding site sequences, using 319 mammalian transcription factor models. Fifty-one and 50 of these models were enriched in distal flanked regions in stimulated HeLa and mouse liver, respectively, relative to randomized sequences, and at least 95% of the distal flanked regions had an exact sequence match for three or more enriched models (Supplemental Figs. TF4, TF6; Supplemental Tables TF1, TF2). For proximal flanked regions, 35 and 34 models were enriched, respectively, and again at least 95% of the regions had an exact match for three or more enriched models (Supplemental Figs. TF5, TF6; Supplemental Table TF2).
These results indicate that the large sets of distal regions that were flanked with H3K4me1, but were not associated with either STAT1 or FOXA2, had sequence properties that were consistent with regulatory regions capable of binding TFs.
Spatial distributions for H3K4me1 and H3K4me3 at distal TF binding sites
We used the high spatial resolution with which we could estimate likely locations of TF binding within a ChIP-seq profile to characterize how associated histone modifications were spatially distributed around TF sites. As noted above, H3K4me1 modifications commonly occurred as a symmetric flanking pair of enriched regions. We used two complementary metrics: read coverage profiles for a ±5-kb region around a maximum enrichment location of TF binding site, and distributions of the distance between such a site and the location of maximal enrichment of its closest modified region. Profiles offered information on how localized and how spatially constrained a mark was; distributions offered information about both spatial constraint and characteristic length scales for modified regions. Profiles were calculated directly from read data without applying a coverage threshold, while distributions were calculated from FDR-thresholded enriched regions.
Coverage profiles for stimulated HeLa cells and mouse adult liver showed that H3K4me3 was more spatially constrained than H3K4me1 (Fig. 3A,B). For both transcription factors, only the coverage profile for proximal H3K4me3 decayed rapidly with distance (Fig. 4), consistent with proximal H3K4me3 being a strong, localized mark for TSS; all other profiles showed coverage that was distributed across the ±5-kb regions characterized.

Spatial relationships for TF-associated H3K4me1 and H3Kme3. Coverage profiles associated with STAT1 in IFNG-stimulated HeLa cells (A) and FOXA2 in mouse adult liver (B). Density distributions for distances between the location of maximal coverage for a region enriched in STAT1 (C) or FOXA2 (D) and the closest H3K4me1- or H3K4me3-enriched region. (Blue lines) H3K4me1; (green lines) H3K4me3; (solid lines) profiles for distal TF regions; (dashed lines) profiles for proximal TF regions. We distinguished these groups using a ±2.5-kb threshold distance from UCSC hg18 or mm8 known gene TSS. The area under each curve was normalized to 1.0.
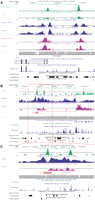
Examples of TF, histone modification, and read mappability profiles. (A) A 15-kb UCSC hg18 genome browser view around the 5′-end of the STAT1 gene (chr2:191,581,501–191,596,500). The gene is transcribed from right to left. Custom tracks are XSET coverage profiles for (from the top) STAT1, H3K4me1, and H3K4me3. Pairs of profiles show IFNG-stimulated cells above and unstimulated cells below. (B,C) Overall 25-kb (chr5:91,517,001–91,542,000) and detailed 8-kb (chr5:91,519,901–91,527,900) UCSC mm8 genome browser views that include a known and a novel enhancer for the albumin (Alb1) gene (Wederell et al. 2008). The gene is transcribed from left to right. ChIP-seq coverage profiles are (from the top) FOXA2, H3K4me1, and H3K4me3 in mouse adult liver. (A–C) The final custom track is a profile for (gray) 27-mer exact match mappability; (black horizontal lines) coverages (heights) that correspond to profile-specific FDR thresholds of ∼0.01 (Table 1).
H3K4me1 distance distributions were similar for distal STAT1 and FOXA2 sites (Fig. 3C,D). Proximal distance distributions for H3K4me1 in stimulated HeLa cells were somewhat broader, consistent with regions close to transcriptional start sites containing dense, complex, and potentially competitive histone modifications; however, in contrast, the distribution for proximal FOXA2 sites was only marginally broader than distal. These distributions suggested that the characteristic distance between a distal transcription factor binding site and H3K4 monomethylation was ∼150 to 200 bp for STAT1 and ∼200 to 300 bp for FOXA2.
Proportions of TF-associated modifications were insensitive to CTCF data
In previous work to characterize histone modifications associated with putative enhancers, distal DHS that are associated with CTCF binding sites or computationally identified motifs were removed because they may mark insulators, rather than enhancers (Barski et al. 2007; Boyle et al. 2008; Wang et al. 2008). To support comparing our TF-based results to these DHS-based results, we assessed whether proportions of H3K4me1/me3 modifications that were associated with distal TF regions were different for regions associated with CTCF sites from published data.
Of distal STAT1 binding sites, 8.7% were within 150 bp (Supplemental Fig. CT1) of one of the ∼12,800 conserved CTCF motifs from ChIP-chip work with primary human fibroblasts (Kim et al. 2007), and an unexpectedly large ∼22% were associated with the ∼38,400 CTCF-enriched regions from T-cells (Barski et al. 2007). Proportions of H3K4me1 and H3K4me3 modifications associated with proximal and distal STAT1 regions were insensitive to whether a region was associated with CTCF (Fig. 2C; Supplemental Fig. CT2).
We did not perform a similar analysis for FOXA2 binding sites, because only 0.8% of the ∼11,000 sites were within 150 bp of the ∼6600 conserved CTCF motifs reported by Kim et al. (2007), and these sites were very weakly associated with Chen et al. (2008)’s ∼64,000 ChIP-seq CTCF locations in mouse ES cells (Supplemental Fig. CT3).
Coverage profiles for H3K4me1 and H3K4me3 around CTCF motifs and enriched regions were generally similar to those around IFNG-stimulated STAT1 and FOXA2 binding sites sites (Fig. 3A,B; Supplemental Figs. CT4, CT5).
These results indicate that, while regions marked by our distal TFs may have included insulators and enhancers, the proportions and coverage profiles for TF-associated histone modifications were insensitive to the presence or absence in the regions of CTCF binding sites or conserved motifs from published data (Barski et al. 2007; Kim et al. 2007; Chen et al. 2008).
Combinations of H3K4me1 and H3K4me3 at STAT1 binding sites were concordant in stimulated and unstimulated HeLa cells
An FDR threshold of ∼0.01 determined ∼20,600 and ∼70,300 STAT1 binding sites in unstimulated and IFNG-stimulated HeLa cells (Table 1). We noted that ∼80% of the STAT1 sites in stimulated HeLa cells were at locations that were associated with H3K4me1 in unstimulated cells (Fig. 1F). Given this striking concordance, we tested whether the location of a STAT1 site in stimulated cells was associated with the same combination of H3K4me1 and H3K4me3 modifications in both stimulated and unstimulated cells. We found that the dominant combinations of marks were highly concordant.
Initially we considered individual histone modifications, without distinguishing whether the enriched regions were associated with STAT1, and noted high levels of concordance. For the ∼301,000 H3K4me1 regions in IFNG-stimulated HeLa cells, concordance rates were ∼67% (random expectation ∼6%) for the default FDR threshold and increased to ∼85% for the most significant 50% of regions (Fig. 1A; Supplemental Fig. MC1A,B). For the ∼76,000 H3K4me3 regions in these cells, concordance rates were ∼55% (random expectation 0.9%), increasing to ∼82% for the most significant 50% of regions (Fig. 1B; Supplemental Fig. MC2A,B).
When we considered STAT1 binding sites, the concordance rate was ∼26% (random expectation 0.9%) for the ∼70,300 stimulated STAT1 regions, increasing to ∼85% for the most significant 50% of regions (Fig. 1C; Supplemental Fig. MC3A,B). Eighty-six percent (random expectation 1.3%) of the ∼20,600 STAT1 binding sites in unstimulated cells were concordant with stimulated sites. The difference in concordance rates reflects the relative number of STAT1 binding sites in stimulated versus unstimulated cells.
We then assessed whether specific combinations of modifications associated with STAT1 binding sites were concordant. As noted above, for the ∼53,000 distal STAT1 sites in stimulated cells, the two dominant combinations of modifications were H3K4me1 without H3K4me3 (39.7%) and H3K4me1 with H3K4me3 (40.5%) (Fig. 2A,C). These patterns were highly concordant. Eighty-four percent of the ∼21,000 distal STAT1 binding sites that were marked by H3K4me1 only in the stimulated state were marked by the same pattern in the unstimulated state, as were 66% of the ∼21,500 sites that were associated with both H3K4me1 and H3K4me3 (Supplemental Fig. PC2).
To characterize pattern concordance in more detail, we divided the distal STAT1 binding sites in stimulated cells into two groups: (1) the 26.7% of locations that were bound in both stimulated and unstimulated cells (Supplemental Fig. PC5), and (2) the 73.3% of locations that were bound in stimulated but not in unstimulated cells (Supplemental Fig. PC7).
Ninety-one percent of locations in the first group were associated with two combinations of modifications: H3K4me1 only (28%), and H3K4me1 with H3K4me3 (63%). Modification patterns associated with these locations were concordant; 84% of locations associated with H3K4me1 only, and 77% of those associated with both H3K4me1 and H3K4me3 were associated with the same pattern in unstimulated cells. The same two H3K4me1/me3 combinations that dominated the first group were associated with 77% of locations in the second group: H3K4me1 only (43%), and H3K4me1 with H3K4me3 (34%). Again, patterns at these distal locations were concordant; 84% of the locations associated with H3K4me1 only, and 61% of those associated with both H3K4me1 and H3K4me3 were associated with the same pattern in unstimulated cells.
These results indicated that both individual histone modifications and specific dominant combinations of H3K4me1 and H3K4me3 modifications were highly concordant between stimulated and unstimulated HeLa cells.
Discussion
In this study, we used ChIP-seq profiles to characterize relationships between H3K4me1 and H3K4me3 modifications that were associated with active distal and proximal regulatory regions. We inferred the regulatory regions from protein–DNA association profiles for two functionally different TFs in two biological systems: STAT1 in untreated and IFNG-treated human HeLa S3 cells, and FOXA2 in mouse adult liver. The TF-based profiles offered a ±50–100-bp spatial accuracy for known functional binding sites and binding motifs for a target TF, and large sets of distal binding sites (∼53,000 for STAT1 in stimulated HeLa cells and ∼9600 for FOXA2) (Robertson et al. 2007; Wederell et al. 2008).
We found that independently derived STAT1 binding sites and histone H3K4me1 regions were strongly associated in HeLa cells, as were FOXA2 binding sites and H4K3me1 regions in mouse adult liver. While a subset of distal TF binding sites in stimulated HeLa cells and mouse liver was associated with both H3K4me1 and H3K4me3, H3K4me1 was a more commonly associated modification, particularly for FOXA2 in the mouse tissue. Furthermore, thresholding histone modification profiles using more stringent FDRs increased the proportion of distal TF sites that were associated with H3K4me1 only, at the expense of sites associated with both H3K4me1 and H3K4me3, suggesting that distal TF regions were associated with more of the H3K4me1 modification than the H3K4me3. Taken together, these results suggest that H3K4me1 is a dominant distal genomic mark for genetic transcriptional regulatory regions and transcription factor binding.
In recent ChIP-seq work with human T-cells, 43% of ∼4000 DHS-based distal regulatory regions were reported as being associated with no significantly enriched histone modifications (Wang et al. 2008). The investigators suggested that such unmarked DHS may have been caused by nucleosome depletion or insufficiently deep sequencing. Our H3K4me1 and H3K4me3 profiles were saturated, and, at profile-specific FDR thresholds of ∼0.01, fewer than 20% of the ∼53,000 distal STAT1 sites in stimulated HeLa cells and the ∼9600 distal FOXA2 regions in mouse liver were associated with neither modification.
Profiles for TFs were unsaturated. Differences between saturation curves for histone modifications and transcription factors may be consistent with a ChIP experiment sampling covalent protein modifications for the histone modifications, and dynamic protein–DNA association equilibria for the TFs (Halford and Marko 2004; Slutsky and Mirny 2004; Benevolenskaya 2007; Agger et al. 2008). Sequencing a histone modification reagent beyond a threshold depth may improve the signal-to-noise ratio for modified sites without revealing new sites; for a transcription factor, deeper sequencing may progressively discriminate sites that have lower affinities, including sites that may not be functional (Li et al. 2008).
Results from both HeLa and mouse liver suggest that a single transcription factor can influence an unexpectedly large fraction of the total genomic repertoire of regulatory elements. In stimulated HeLa cells, 24.6% of ∼301,000 H3K4me1 regions were associated with STAT1 binding; in adult mouse liver, 5.5% of ∼227,000 H3K4me1 regions were associated with FOXA2 binding. The fourfold difference may reflect the different roles of the two transcription factors. STAT1 is involved in acute responses to a signal that rapidly modifies transcription (Brierly and Fish 2005). In contrast, FOXA2 has been proposed as a pioneer factor and is important in both in establishing gene expression patterns during tissue development and in regulating homeostasis in the adult (Wederell et al. 2008). We have previously shown that distal and proximal STAT1 and FOXA2 regions correlate closely with known functional sites, and with expected DNA sequence motifs (i.e., STAT1-like or FOXA2-like) (Robertson et al. 2007; Wederell et al. 2008).
Mammalian genomes encode at least 2000 DNA binding proteins (Wilson et al. 2008), and we anticipate that ChIP-seq experiments using antibodies for other TFs would identify binding sites in different subsets of the distal H3K4me1-marked regions than we found associated with STAT1 and FOXA2 in the current work. Consistent with this, we demonstrated that a majority of the large number of distal regions that were symmetrically flanked by H3K4me1 had sequence properties that were expected for regulatory regions. From these results, we estimate that the human and mouse cell types in this study contain at least 100,000 distal regulatory regions that are symmetrically marked by H3K4me1.
We characterized general parameters for spatial relationships between TF binding sites and methylated regions using coverage profiles relative to a TF region maximum and distributions of distances between the maximum of a TF region and that of its nearest modified region. Coverage profiles suggested that the characteristic distance between distal transcription factor binding and H3K4 monomethylation was between ∼200 and 350 bp, while distance distributions indicated ∼150 to 200 bp for STAT1 and ∼200 to 300 bp for FOXA2. These lengths suggest a preferred pattern in which one or more nucleosomes adjacent to the transcription factor binding site may have other histone modifications but are not mono-methylated. Alternatively, transcription factor binding may be accompanied by substantial changes in nucleosome positions.
Recent results have associated specific histone modifications with two classes of poised regions: bivalent chromatin domains in ES cells and poised polymerase at inactive promoters (Mendenhall and Bernstein 2008). We noted that a large majority of sites that bound STAT1 in HeLa cells did so only in stimulated cells, and that the large majority of these binding locations were associated with the same combinations of H3K4me1 and H3K4me3 modifications in both unstimulated and stimulated cells. Some of these regions may have been prepared by regulated processes to allow rapid high-affinity binding of activated STAT1 dimers and trimers following IFNG stimulation. At the same time, we anticipate that the majority of the H3K4me1-associated regions that we determined may not have been poised in this way, but were likely occupied by other transcription factors and cofactors required for ongoing cellular maintenance and homeostasis.
Although H3K4me1 was the dominant distal mark, being associated with ∼80% of distal transcription factor binding sites in both HeLa cells and mouse adult liver, we also found that ∼50% of sites marked with either H3K4me1 or H3K4me3 in IFNG-stimulated HeLa cells were associated with both H3K4me1 and H3K4me3, consistent with the findings reported for T-cells (Barski et al. 2007; Wang et al. 2008). However, in the mouse tissue, far fewer distal regulatory elements were marked by H3K4me3, and ratios of associated H3K4me1 and H3K4me3 more closely resembled the H3K4me1-only model originally proposed from ENCODE region data (Heintzman et al. 2007). These results suggest that modifications associated with distal binding sites may vary for functionally different transcription factors or between immortalized cells and tissues. Surveying other tissues and cellular states for histone modifications associated with transcription factor binding, and including transcriptome data, should further clarify distal regulatory regions and the functional relevance of the associated histone modifications in human and mouse genomes.
Methods
Availability
ChIP-seq data are available from http://www.bcgsc.ca/data/histone-modification.
The enrichment profiles in Figure 4 were generated with FindPeaks v3.1.9.2 (Fejes et al. 2008), which is available at http://www.bcgsc.ca/platform/bioinfo/software/.
Chromatin immunoprecipitation
HeLa S3 cells
STAT1 ChIP samples were prepared from IFNG-stimulated and unstimulated HeLa S3 cells. Cultures of 5 × 108 HeLaS3 cells were either induced with 5 ng/mL human recombinant IFNG (R&D Systems), for 30 min at 37°C, 5% CO2; or left untreated. For STAT1, ChIP experiments were performed as previously described (Robertson et al. 2007). For H3K4me1 and H3K4me3, ChIP experiments were performed as follows: Cells were cross-linked with 1% formaldehyde for 10 min at room temperature. Cross-linking was stopped by the addition of glycine to 125 mM final concentration, and cells were washed twice in cold 1× Dulbecco’s PBS. Cells were resuspended in 3 mL of ChIP lysis buffer (50 mM Tris-HCl at pH 8.0, 1% SDS, 10 mM EDTA, 1 complete protease inhibitor cocktail tablet [Roche]), incubated for 30 min at 4°C before being passed six times through a 1-mL syringe, and collected by centrifugation at 5000 rpm for 10 min at 4°C. After the supernatant was removed, the pellet was resuspended in 750 μL of ice-cold ChIP lysis buffer. Samples were sonicated in three 250-μL aliquots at amplitude 7 using a Fisher Scientific Model 550 Sonic Dismembrator (Fisher) for a total sonication time of 10 min to produce chromatin fragments of 0.5 kb on average. After centrifugation and quantification, the chromatin fractions were pre-cleared for 2 h at 4°C with 120 μL of a 50% slurry of protein A/G-Sepharose beads (Amersham). These beads were prepared by two washings in IP buffer (10 mM Tris-HCl at pH 8.0, 1% Triton X-100, 0.1% deoxycholate, 0.1% SDS, 90 mM NaCl, 2 mM EDTA, 1 Complete EDTA-free protease inhibitor cocktail tablet [Roche]) and a 3-h incubation with 75 μg of sonicated salmon sperm DNA and 200 μg of BSA per mL of solution in IP buffer. The beads were then resuspended 1:1 in IP buffer and used for ChIPs. Each immunoprecipitation was carried out for 1 h at 4°C with 100 μg of pre-cleared chromatin and 2.5 μg of anti-histone H3 monomethyl K4 (Abcam) or anti-histone H3 trimethyl K4 (Abcam). Complexes were recovered by overnight incubation at 4°C with 20 μL of the protein A/G-Sepharose beads solution. Precipitates were washed twice with ChIP wash buffer (20 mM Tris-HCl at pH 8.0, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl) and once with ChIP final wash buffer (20 mM Tris-HCl at pH 8.0, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl). Precipitates were resuspended in 100 μL of elution buffer (100 mM NaHCO3, 1% SDS) with 5 μg of freshly added RNase (DNase free; Roche) and incubated for 2 h at 68°C with shaking in a thermomixer to reverse the cross-link. The beads were pelleted by centrifugation, and the supernatant was collected. Elution was then repeated with the addition of 100 μL of Elution Buffer and incubation for 5 min at 68°C. After pooling, the DNA was recovered from the eluate using the QIAquick PCR Purification kit (QIAGEN) according to the manufacturer’s instructions. ChIP-seq libraries were constructed as described (Robertson et al. 2007) using 147 ng and 113 ng of IP’ed DNA for the unstimulated and stimulated HeLa S3 samples, respectively.
Adult mouse liver
FOXA2, H3K4me1, and H3K4me3 immunoprecipitations were conducted as outlined previously (Wederell et al. 2008). Briefly, adult female C57Bl/6J mouse livers were fixed for 10 min with 1% formaldehyde after homogenization. Following quenching with glycine, cells were pelleted, washed, and lysed, and then nuclei were pelleted and then resuspended in nuclear lysis solution. Nuclei were sonicated in ice water for 20 cycles of 30 sec (Sonicator 3000; Misonix). Debris was spun down, and the 60 μg of fragmented chromatin was precleared for 1 h at 4°C with 100 μL of Protein G agarose beads (Active Motif), 0.5 μL of Protein Inhibitor Cocktail (Active Motif), and ChIP dilution buffer. Supernatants were removed from the beads following centrifugation and transferred to siliconized tubes to which 3 μg of anti-FOXA2 [HNF-3β (M-20), sc-6554; Santa Cruz], anti-histone H3 monomethyl K4 (Abcam), anti-histone H3 trimethyl K4 (Abcam), or normal rabbit IgG (Santa Cruz) was added. At the same time, fresh protein G agarose beads were preblocked with BSA and herring sperm DNA. Following overnight incubation at 4°C, chromatin-antibody reaction was added to the blocked beads and incubated for 4 h rocking at 4°C. The beads were then precipitated by centrifugation and washed as follows: low salt buffer, high salt buffer, lithium chloride buffer, and twice with TE buffer. DNA–protein complexes were eluted twice with 125 μL of fresh elution buffer and rotation for 15 min at room temperature. To reverse the cross-linking, 5 M NaCl was added to the eluted immunoprecipitated complex to a final concentration of 0.192 M, and it was incubated overnight at 65°C. Protein was excluded from the DNA fragments by treatment with 0.5 μL of 20 μg/μL proteinase K (Invitrogen) along with RNaseA (Sigma) for 2 h at 50°C. The DNA was purified by two rounds of phenol-chloroform extraction and ethanol precipitation and resuspended in 50 μL of dH2O.
Aligning sequence reads to reference genomes
Sequence reads of 27 bp or 32 bp derived from Illumina 1G sequencers were aligned to the NCBI reference human (hg18) and mouse (mm8) genomes using Eland (Illumina), which allows a uniquely aligned read to have up to two mismatching bases. Because the global sequencing error rate tended to be higher in later sequencing cycles, we maximized the number of aligned reads by using an iterative alignment approach; any read that did not align uniquely to the genome was shortened by removing several terminal base pairs and was realigned using Eland, to a minimum read length of 23 bp (Robertson et al. 2007). Only sequence reads that aligned to unique genomic locations were retained.
Filtering reads
We removed from Eland output files records for any aligned read whose sequence contained uncalled bases, as well as records for reads whose sequences were similar to sequences for gel size selection “ladders” or sequencing adapters, for which we allowed up to four base mismatches. While we expected that a deeply sequenced data set could contain a low rate of multiple sequence reads that corresponded to the same DNA fragment start, such “duplicate reads” could also arise from nonlinearities in the library preparation and cluster generation processes. Given these potential artifacts, we conservatively collapsed, into a single read all sets of multiple reads that corresponded to a single DNA fragment start.
Identifying enriched regions
We used FindPeaks v2.0 to generated overlap profiles for immunoprecipitated and nonspecific DNA fragments by extending directional reads to the estimated mean fragment length, creating virtual fragments (XSETs), then profiling the number of overlapped XSETs across the genome (Robertson et al. 2007; Wederell et al. 2008). We estimated the mean DNA fragment length as 200 bp from the gel size separation step in library preparation. We used distributions of distances of directional sequence reads from enriched STAT1 locations to confirm the longest fragment lengths (Fejes et al. 2008) and verified that the number of significant enriched regions identified was relatively insensitive to the XSET length estimate in the range of 100 to 300 bp (data not shown).
We identified enriched regions as islands of continuously overlapping XSETs that were separated by gaps from other islands (Wederell et al. 2008). Taking an island’s height as its maximum overlapped XSET count, a raw profile contains both singleton XSETs and XSET islands with a wide range of heights. For each experimental data set, we computationally estimated a relationship between island height and false discovery rate (FDR), using an extension of the approach described previously (Robertson et al. 2007). We assumed that every aligned read belonged to one of two classes: signal, due to specific protein–DNA binding, and background. For the background, we assumed the Poisson model described in Lander and Waterman (1988) and calculated the number of islands, K, expected at random for a given number of reads in the island, n, using K(n) = NBA2( 1− A)(n − 1), with A = exp(−lNB/L). In these equations, NB is the number of background reads, that is, reads due to nonspecific protein–DNA binding; L is the “mappable” (effective) genome length, that is, the overall fraction of a genome length to which reads can be aligned to unique locations; and l is the XSET length. After setting the XSET length as noted above, we used the above equations to calculate values for NB and L, assuming, given our sequencing depths, that all islands that contained one or two reads represented background. Next, we used Monte Carlo simulation to model various island configurations and calculated the distribution of island heights (h) for a given number of reads, M(h;n). By combining K(n) and M(h;n), we obtained the expected number of randomly generated islands as a function of island height. The empirical FDR for a particular island height threshold was the ratio between the number of simulated islands and the number of experimentally observed islands for that threshold. We used the experimental proportions of aligned read lengths (between 23 and 32 bp) and genome read mappability resources (see below) to confirm the effective genome length calculated in the above approach for both human and mouse. For single-end reads in this length range, the effective lengths were ∼70% of the respective nominal genome lengths, or ∼85% of the finished genome lengths.
For STAT1, FOXA2, and H3K4me1, we retained only islands that were at least as tall as a threshold height that corresponded to an FDR of ∼0.01. We then refined islands into narrower regions by trimming each island at a fraction of its height (typically 20%). The trimming operations removed low-height flanks and often delivered separate regions that had been linked by low-height profile bridges (see Fig. 2).
For the work reported, we used current filtering, profiling, and thresholding methods to reanalyze the published STAT1 and FOXA2 data (Robertson et al. 2007; Wederell et al. 2008).
Sequencing depth and saturation
We estimated whether the sequencing depth, that is, the number of uniquely aligned reads, was sufficient to identify essentially all enriched regions that were available under the conditions of an experiment, that is, whether the experiment had “saturated,” or completely represented, the available regions (Robertson et al. 2007). Given a set of aligned reads from an experiment in a filtered Eland output file, we numerically simulated experiments that generated fewer reads by computationally sampling random subsets of aligned reads from the file and calculating the number of FDR-thresholded enriched regions for each subset. The simulations used the estimated effective mappable genome lengths (above) and were repeated five times at each sequencing depth. We inferred a relationship between the number of significant regions and the number of sequence reads, using a set of subsampling experiments (Supplemental Fig. SA1). A profile plotted to represent such relationships shows the number of enriched regions at a constant FDR value of ∼0.01. Region counts at this FDR were linearly interpolated from the counts for the two flanking FDR values, which corresponded to integer-valued region heights. Interpolated region counts were repeatable; in all cases, ranges of values over five simulations fell within the circular plot symbols. Error bars show average flanking region counts for five simulations. For each experiment, we reported an FDR thresholded number of enriched regions that corresponded to the FDR nearest to the target value of 0.01.
Sequence read mappability
In this study, Illumina sequence reads were 27 or 32 bp, and we generated enrichment profiles using only the reads that Eland aligned (or mapped) to unique genomic locations. Because we mapped reads iteratively, the lengths of mapped sequence reads from each experiment ranged from 23 to 32 bp. Reference genome sequences are partially repetitive, and a longer sequence read typically can be mapped uniquely to a larger fraction of a genome sequence. ChIP-seq profiles in genomic regions that have a uniformly high mappability can be interpreted as potentially reflecting biological processes; conversely, no enriched regions are expected in genomic regions that have consistently low mappability, and enriched region profiles need to be interpreted with care in genomic regions that have variable read mappability. To address this, we exhaustively enumerated hg18 and mm8 reference genome sequences to generate genome-wide mappability data resources that profiled the extent to which short DNA sequences could be uniquely aligned to the respective genomes. We used these resources to assess enriched region profiles in targeted genomic regions using low- and high-throughput approaches. Figure 4 shows examples of mappability profiles and illustrates two points. First, because genomic annotations for repetitive regions need not correspond to low exact-match mappabililty, reads can be mapped into certain repeat types. Second, using XSETs makes it possible to assign coverage across some low mappability regions, albeit with attenuated signal strength. For high-throughput approaches, we assessed 27-mer mappability profiles for ±3-kb genomic regions around the maximum of each STAT1 (IFNG) region (Supplemental Material, section MP). We considered profiles for STAT1 regions whose maximum did not overlap an H3K4me1 region and divided the profiles into four subgroups: STAT1 regions without an associated H3K4me1 region, with an H3K4me1 region only on one side but not on the other, and with an H3K4me1 region on both sides. We determined association (or lack of association) between STAT1 and H3K4me1 regions with a 1-kb threshold distance between the locations of maximum coverage for a transcription factor region and an H3K4me1 region.
Acknowledgments
We thank P. Bickel, B. Brown, E.H. Margulies, and G.K. McEwen for making their implementation of the block bootstrap method available to us. S.J.M.J., P.A.H., and M.A.M. are scholars of the Michael Smith Foundation for Health Research. Funding for this work was provided in part by Genome Canada, Genome British Columbia and the Canadian Institute of Health Research.
Footnotes
-
↵5 Corresponding author.
↵5 E-mail sjones{at}bcgsc.ca; fax (604) 876-3561.
-
[Supplemental material is available online at www.genome.org. The ChIP-seq data are available from www.bcgsc.ca/data/histone-modification.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.078519.108.
-
- Received March 17, 2008.
- Accepted September 4, 2008.
-
Freely available online through the Genome Research Open Access option.
- Copyright © 2008, Cold Spring Harbor Laboratory Press








