
Pathogen corruption and site-directed recombination at a plant disease resistance gene cluster
Abstract
The Pc locus of sorghum (Sorghum bicolor) determines dominant sensitivity to a host-selective toxin produced by the fungal pathogen Periconia circinata. The Pc region was cloned by a map-based approach and found to contain three tandemly repeated genes with the structures of nucleotide binding site–leucine-rich repeat (NBS–LRR) disease resistance genes. Thirteen independent Pc-to-pc mutations were analyzed, and each was found to remove all or part of the central gene of the threesome. Hence, this central gene is Pc. Most Pc-to-pc mutations were associated with unequal recombination. Eight recombination events were localized to different sites in a 560-bp region within the ∼3.7-kb NBS–LRR genes. Because any unequal recombination located within the flanking NBS–LRR genes would have removed Pc, the clustering of cross-over events within a 560-bp segment indicates that a site-directed recombination process exists that specifically targets unequal events to generate LRR diversity in NBS–LRR loci.
Most pathogen recognition and resistance in plants is orchestrated by proteins that contain a nucleotide binding site (NBS) and leucine-rich repeats (LRRs). NBS–LRR-based resistance reactions require direct or indirect interaction with a pathogen-encoded effector molecule (itself a virulence factor) that initiates a signal transduction process leading to resistance. This resistance often involves a hypersensitive reaction leading to an organized destruction of plant tissues around the site of infection (Greenberg 2001). The NBS–LRR resistance genes commonly act against biotrophic pathogens, microbes that require metabolically active plant tissues to produce a productive infection. NBS–LRR genes are abundant in plants, with different genes acting against different pathogens and races of pathogens. Hence, this type of resistance is also called race-specific resistance. The race specificity of NBS–LRR genes in plants appears to be determined by effector interaction with the solvent-exposed LRR domains (Jones and Jones 1997; Dodds et al. 2001).
Much less is known about plant diseases associated with toxin production. The toxin-producing organisms, commonly fungi, require a somewhat debilitated host to infect and/or proliferate extensively. The molecular structures and origins of toxins can be quite different, as are the dominant or recessive single genes in the host that provide resistance (Wolpert et al. 2002).
The root and crown rot of sorghum known as milo disease is caused by the peritoxin produced by the saprophytic fungus Periconia circinata (Leukel 1948). The toxin alone mimics most symptoms of the disease, including highly condensed heterochromatin and efflux of potassium ions, traits reminiscent of the hypersensitive response (Arias et al. 1983; Dunkle and Macko 1995).
The dominant susceptibility gene for milo disease, Pc, naturally mutates to the resistant pc allele at a rate of about one per 8000 gametes (Schertz and Tai 1969). This high level of instability is unidirectional: pc-to-Pc mutations have not been observed. The Pc gene was mapped to a 0.9-cM region on the short arm of sorghum chromosome 9 and physically localized to a 110-kb segment in inbred BTx623 (pc/pc) that was sequenced (Nagy et al. 2007). Sequence annotation revealed 12 genes in the region. Each of these 12 gene candidates was analyzed in susceptible (Pc/Pc) and 13 spontaneous (i.e., isogenic) resistant (pc/pc) derivatives (M1–M13) using the mutation detection system called single-stranded conformational polymorphism (SSCP). All gene candidates were identical between the Pc/Pc and pc/pc plants, except a tandemly duplicated NBS–LRR gene family that exhibited rearrangement in all 13 mutants (Nagy et al. 2007). This study suggested that one or more of the NBS–LRR genes was the Pc locus, although it could not exclude the possibility that Pc was some other unidentified gene present between the NBS–LRR loci and lost by unequal recombination in pc (resistant) progeny like the sequenced BTx623 haplotype. In this work, we identify the Pc gene and precisely describe the molecular rearrangements that gave rise to Pc-to-pc mutations.
Results
A sorghum fosmid library was constructed from Pc/Pc cultivar Colby. Four clones covering the NBS–LRR genes were sequenced and assembled into a single contig of 78,705 bp (NCBI accession no. EU583216). Annotation revealed three NBS–LRR genes (A, B, and C), arrayed tandemly in a head-to-tail fashion (Fig. 1; NCBI accession nos. ACB72454, ACB72455, and ACB72456). The three paralogs are predicted to encode proteins of 1277, 1194, and 1203 amino acids, respectively. The paralogs are separated by respective A–B and B–C intergenic regions of 12,638 and 13,713 bp. These intergenic regions in Pc/Pc Colby contain large retrotransposon insertions that are homologous to each other (92% sequence identity) and to those in the pc region of inbred BTx623 that was sequenced earlier (Nagy et al. 2007; NCBI accession no. EU810765). Their degree of sequence identity and common location/orientation indicate that the two retroelements are the products of a single insertion that was subsequently duplicated (along with Pc paralogs) by unequal recombination. The N-terminal and NBS regions (bp 1–1399) are identical in paralogs A and C, while paralog B is different from the other two across the entire gene (Supplemental Fig. S1). Among the many sequence differences between A, B, and C, a 75-bp insertion and a 141-bp deletion in the B coding region relative to A and C are particularly prominent. These sequence changes, however, leave each of the genes in the same reading frame, so a functional protein is possible. Aside from the indels, the overall nucleotide similarity was high (>90%) in all pairwise comparisons. Sequencing of RT-PCR products revealed that all three paralogs were transcribed in the seedling roots of uninfected Pc/Pc Colby plants.

Annotation of the Pc region in sorghum cultivar Colby as shown in an Apollo output diagram. The annotation results are shown in the black fields. Yellow blocks represent the gene homologies identified by BLASTX; pink blocks signify the predicted genes (FGENESH). The names below the identified homologies indicate the proteins that are predicted to be encoded by these candidate genes. The Pc gene is the central member of the NBS–LRR gene triplet.
In a previous study, 13 independent Pc-to-pc mutations were identified as natural events in the self-cross progeny of a line called Pc/Pc Colby (Schertz and Tai 1969; Nagy et al. 2007). These mutations, M1–M13, are thus true isolines with their Pc/Pc Colby parent. Long-distance PCR was used to amplify the paralogs in the M1–M13 (Pc mutant) genotypes. Seven of the 13 mutants (M1–M7) contained a single paralog that was identical with paralog A in the 5′ region, while their 3′ end aligned with paralog C. These are the predicted products of unequal recombination events that occurred between paralogs A and C (Fig. 2). RT-PCR studies indicated that the A/C paralog is expressed in the seedling roots of all seven mutants (Fig. 3).
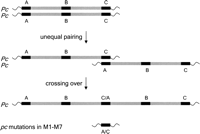
Unequal recombination events between paralogs A and C in Pc mutants M1–M7.

Expression analysis on susceptible Colby (Pc) and nine derived pc isolines using RT-PCR. A primer pair common to all three NBS–LRR paralogs was used. Negative controls (C.), lacking reverse transcriptase in their reaction mixtures, were also included. The PCR products were cloned and sequenced to identify expressed paralogs.
Three of the mutants (M8–M10) exhibited a single paralog that was indistinguishable from paralog C. Because A and C paralogs are identical in sequence from bp 1399 within the genes to some 870 bp upstream of the genes, we cannot precisely map where these unequal recombination events occurred. The single NBS–LRR paralog in these mutants was also found to be expressed (Fig. 3).
Mutant M11 contained intact paralogs A and C but lacked paralog B. An unequal recombination event between the A–B and B–C intergenic regions can explain this result. These two intergenic regions are highly similar to each other (76%), including many long (several hundred base pairs) stretches of identical DNA that would provide ample homology for unequal recombination. RT-PCR analysis demonstrated that paralogs A and C in M11 were also transcribed. Therefore, the only obvious genetic change in M11 was the loss of paralog B, suggesting that it is the Pc locus.
Mutant M12 was found to contain intact paralogs A and C and a paralog B that is truncated by an internal deletion of 468 bp in the LRR region. The deletion breakpoints are flanked by 8-bp homologous motifs (GTCTTTAA) suggestive of illegitimate recombination (Gorbunova and Levy 1999; Bennetzen 2007; Wicker et al. 2007). All three paralogs in M12 were found to be transcribed. These results demonstrate that internal deletion in paralog B was sufficient to cause a Pc-to-pc mutation, thereby proving that paralog B is the Pc gene.
Mutant M13 carries an intact paralog A and a recombinant B/C paralog. RT-PCR studies using gene-specific primers indicated that paralogs A and B/C are both transcribed, but the B/C paralog is only weakly expressed (data not shown). The molecular origin of this haplotype required an unequal recombination localized in the LRR regions of paralogs B and C, but the low level of expression from this recombined B/C is unexplained.
All of the predicted rearrangements and RT-PCR results were retested using gene-specific or rearrangement-specific primers. The genomic PCR studies confirmed all of the predicted rearrangements by showing that (1) in all cases an intact gene B was missing and (2) the predicted chimeric gene (A/C or B/C) was present (data not shown).
Thirteen rearrangements, 12 unequal recombinations and one deletion, were detected in the 13 Pc-to-pc mutations (Table 1). Eight of these unequal recombinations were resolved inside a 560-bp segment of the LRR region (bp 3062–3622). This segment was favored for unequal recombinations, in spite of the fact that it is less homologous than other regions (Fig. 4) in the Pc homologs. The deletion in M12 caused by illegitimate recombination also occurred within this segment, suggesting an inaccurately repaired double-strand break inside this high-recombination region (Kirik et al. 2000; Ma and Bennetzen 2006).
Rearrangement of NBS–LRR paralogs A, B, and C in Pc mutant isolines
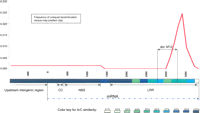
Distribution of unequal recombination events along the consensus sequence of the NBS–LRR paralogs A and C (3737 bp) and their upstream noncoding regions (870 bp). Similarity between paralogs A and C is shown using a color-coded bar below the distribution graph. Seven of the 11 A–C recombinants and the single B/C recombinant (M1–M7, M13) were localized in a highly variable segment of the LRR region. Recombination frequency in this region was significantly higher (P < 0.0005) than the overall frequency measured along the complete gene. A 468-bp internal deletion in paralog B of M12 overlaps with the A/C recombination hot-spot, suggesting that an imprecisely repaired double-strand break also occurred in the high-recombination region.
Discussion
In plants, the major role found for NBS–LRR genes is their involvement in the pathogen-recognition process leading to disease resistance. The results for Pc indicate that an NBS–LRR gene, paralog B, can also be corrupted by a necrotrophic pathogen to create disease susceptibility. A similar result is predicted for the Vb toxin susceptibility gene of oats. At this locus, susceptibility to the toxin victorin produced by the necrotroph Cochliobolus victoriae, has not been separable from resistance to the biotroph Puccinia coronata conditioned by Pc-2. Any mutations creating resistance at Vb by its inactivation also creates susceptibility to specific races of P. coronata (Mayama et al. 1995). Recent studies, using Arabidopsis as a surrogate, suggest that victorin causes host susceptibility by inducing a hypersensitive resistance reaction that allows the necrotroph to penetrate host tissues (Lorang et al. 2007). Hence, as also seen in the innate immunity of animal cells associated with TLR (Toll/interleukin 1 receptor domain, LRR motif) genes, some pathogens have mechanisms to utilize modes of host resistance to create enhanced pathogen susceptibility (Akira et al. 2006). In this regard, it is likely that the Pc locus provides some positive contribution (for instance, resistance to a fungal biotroph) in its land of origin, Africa. This predicted pathogen, or the appropriate avirulence gene target, is apparently lacking in the US, because most or all commercial sorghum lines have been selected to carry pc alleles to avoid milo disease.
In this study, we were unable to identify which unique amino acid variants, or combination of variants, at gene B are responsible for its function as Pc, that is, sensitivity to the P. coronota peritoxin. From analogy with other studies (Jones and Jones 1997; Dodds et al. 2001), it is likely that the LRR component will be responsible for the recognition process. Gene B does have the most different structure because of two major indels, one in the LRR region, compared with paralogs A and C. Hence, it is tempting to speculate that Pc function will be derived from “B-unique” variations in the LRR domains, like the 141-bp indel or the respective leucine and serine amino acids at positions 918 and 941. However, because only one of the Pc-inactivational recombinations was between B and either paralog (M13, and this one may yield a pc phenotype because of a low level of subsequent expression), it is not possible to identify specific determinants of Pc toxin recognition. The comparison to the pc/pc haplotype of BTx623 sequenced earlier was also of little value for this purpose, because the origin of this haplotype is unknown and because it shows many widely scattered variations between its three Pc-like genes and the Pc gene of Pc/Pc Colby.
Eight of the 12 unequal recombinations and an illegitimate recombination were detected inside a 560-bp segment of the LRR region (bp 3062–3622) that was less homologous than the other regions. The higher polymorphism of the LRR domains is generally viewed as a result of diversifying selection for novel pathogen recognition properties, whereas the more homologous NBS domains are an expected result of purifying selection on a highly conserved signal transduction role (Michelmore and Meyers 1998; Palomino et al. 2002). A recent study (Wicker et al. 2007) has suggested that illegitimate recombination may employ low levels of LRR homology, which create short tandem duplications that can lead to further LRR amplification by unequal homologous recombination or replication slippage. The current Pc results, however, indicate that unequal homologous recombination events are often resolved within relatively low homology LRR repeats, thus indicating that many of these small LRR copy number changes could originate by unequal homologous recombination as well.
Given that paralog B in the Pc region is the actual Pc gene, it was surprising that more A/B or B/C chimeric genes were not created to remove Pc function. These were expected, and it was hoped that they would delimit the components of B that are responsible for peritoxin recognition. Their near-absence suggests that gene B is poorly aligned for recombination, that the peritoxin recognition component of gene B is redundant (e.g., at both its N-terminal and C-terminal ends), or that the A/B and B/C chimerics have some unknown negative biological outcome. We believe that the latter possibility is most likely, and such an outcome could be the creation of a lesion mimic (Hu et al. 1996) or other debilitating genetic function.
Previous studies of resistance gene evolution have suggested that most NBS–LRR gene clusters in plants exhibit rare but important unequal homologous recombination as a mechanism for changing gene number and possibly creating new resistance gene specificities (Michelmore and Meyers 1998). However, some complex loci, like Rp1 of maize (Bennetzen et al. 1988; Richter et al. 1995) and Pc of sorghum (Schertz and Tai 1969), create new specificities at a very high rate by unequal homologous recombination. In haplotype analyses, many of these unequal recombination events map within LRR arrays (Parniske et al. 1997; Meyers et al. 1998), but this was thought to be a likely outcome of rare LRR-sited events that persisted because of selection for new haplotypes that originated a new specificity for pathogen race recognition. However, in this Pc study, only loss of paralog B was required to create the Pc-to-pc phenotype that was selected. Neither the plants nor the gametes were subjected to any other selection, such that all progeny of the pc mutant-isolation cross survived. Hence, these results indicate a preferential site-direction of the cross-over resolution of recombination events to a small area within an LRR cluster, and one that is particularly low in sequence homology. This phenomenon has the earmarks of site-directed recombination (albeit directed to a small region rather than precise nucleotides). Site-directed recombination is a rare eukaryotic phenomenon not previously observed in plants but one that has key roles in other eukaryotic kingdoms (Haber 1998), including in the creation (Maizels 2005) or escape (Taylor and Rudenko 2006) of a disease resistance response.
Methods
Construction of the fosmid library and sequence analysis
Genomic DNA was isolated from sorghum cultivar Colby using the standard CTAB method (Murray and Thomson 1980). For fosmid library construction, the CopyControl Fosmid Library Production Kit from Epicentre was used according to the manufacturer’s instructions. Library screening of clone pools and superpools was carried out using PCR primers specific for the NBS–LRR gene family. These primers were designed using the corresponding genomic region in sorghum line BTx623 as a template, sequenced in a previous study (Nagy et al. 2007). Four positive clones were shotgun subcloned using the TOPO TA Cloning Kit (Invitrogen) and sequenced.
For base calling and sequence assembly, the programs phred and phrap were used, respectively (Ewing et al. 1998). Contiguous sequences (contigs) were visualized and edited using the program consed (Gordon et al. 1998). Sequence homology searches were performed using the BLAST program package (Solovyev and Salamov 1997). For gene prediction, the program FGENESH (Altschul et al. 1990) was applied. The annotation results were edited and displayed with Apollo software (Lewis et al. 2002).
Analysis of gene configurations in the Pc-mutant isogenic lines
Two primers, PallF (GAACATTTCTGCCGCCACATTTC) and PallR (AGCAGTTAGGCGTTGTATGGATTG), common to the termini of all three paralogs were used to amplify the NBS–LRR units in the Pc-mutant isolines. The long-distance (LD) PCR mixture (50 μL) contained 150 ng of genomic DNA, 2.5 U of Herculase Enhanced DNA polymerase (Stratagene), 100 ng of each primer, and 200 μM of each dNTP. The thermocycling profile was set up according to the instructions provided with the DNA polymerase. The PCR products were isolated from an agarose gel using the Qiaex II Gel Extraction Kit (QIAGEN) and cloned using the TOPO TA Kit. At least 24 of the clones were sequenced from each genotype to reconstitute their gene configurations. Sequence alignments between the susceptible Colby and Pc-mutant isoline NBS–LRR genes were performed using ClustalW (Thompson et al. 1994). Alignments were viewed and edited with Mega 3.1 (Kumar et al. 2004).
The gene configurations in the Pc mutants were confirmed using PCR primers specific for each of the three paralogs (Supplemental Table S1). All primer-pairs flanked the high-recombination region. The primers were used in various combinations that allowed the amplification of the wild-type (A, B, and C) and the recombinant (A/C or B/C) paralogs. Wherever it was possible, multiple primer-pairs were used for confirmation (Supplemental Table S1). The 20-μL PCR mixture included 1.5 mM MgCl2, 0.2 mM of each dNTP, 8 pmol from each of the forward and reverse primers, 0.6 U Taq polymerase (Promega), and 1 ng/μL genomic DNA. Touch-down PCR conditions were employed to avoid nonspecific amplifications. The annealing temperature decreased gradually from 64°C to 58°C through seven cycles, followed by 33 cycles at 58°C.
Expression analysis of the NBS–LRR paralogs
Sorghum seeds were germinated in sterile MS medium on filter paper, and total RNA was isolated from 2- to 5-cm roots of seedlings using the TRIzol reagent (Invitrogen). The mRNA fraction was purified with the FastTrack 2.0 mRNA isolation Kit (Invitrogen). cDNA was synthesized using the SuperScript III First-Strand Synthesis System (Invitrogen). A primer pair (rtF: ATCATCATC TGGCTGGGAAC, rtR: AACCAGGGCAACCATAAATG) that was common to all three paralogs was designed and used for PCR. The expected sizes of the PCR products were 431 bp for paralogs A and C and 290 bp for paralog B (Fig. 3). The cDNAs and their corresponding negative controls having no reverse transcriptase in the reaction mixture were used as a template. The resulting bands were isolated from the gel and cloned with the TOPO TA Cloning Kit. Twenty-four clones were sequenced from susceptible Colby, and each of the mutant derivatives carrying multiple paralogs. Twelve clones were sequenced from mutants containing a single paralog. The expression of paralog B/C in the mutant M13 was not detected using primers that would amplify all paralogs (Fig. 3); therefore, primers specific for the B/C paralog were used in RT-PCR to confirm its expression (Supplemental Table S1). The PCR conditions were as described above for the paralog nonspecific markers.
Statistical analysis of the distribution of unequal recombination events
The frequency of recombinations between the A and C paralogs was calculated for each interval flanked by single nucleotide polymorphisms. The number of recombinations was divided by the length of the corresponding interval. The frequency data were averaged for every 100 base pairs along the A–C consensus sequence. The mean recombination frequency for the total length was compared with the one calculated for the recombination hot-spot by using the Student's t-test.
Acknowledgment
This work was supported by a grant from the U.S. Department of Agriculture (grant no. 20063531917462).
Footnotes
-
↵1 Corresponding author.
↵1 E-mail maize{at}uga.edu; fax (706) 542-3910.
-
[Supplemental material is available online at www.genome.org.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.078766.108.
-
- Received March 21, 2008.
- Accepted August 13, 2008.








