

Summary
In this study, we used whole genome sequencing and gene expression profiling of 215 human induced pluripotent stem cell (iPSC) lines from different donors to identify genetic variants associated with RNA expression for 5,746 genes. We were able to predict causal variants for these expression quantitative trait loci (eQTLs) that disrupt transcription factor binding and validated a subset of them experimentally. We also identified copy number variant (CNV) eQTLs, including some that appear to affect gene expression by altering the copy number of intergenic regulatory regions. In addition, we were able to identify effects on gene expression of rare genic CNVs and regulatory single nucleotide variants, and found that reactivation of gene expression on the X chromosome depends on gene chromosomal position. Our work highlights the value of iPSCs for genetic association analyses and provides a unique resource for investigating the genetic regulation of gene expression in pluripotent cells.
Graphical abstract
Introduction
Since their discovery 10 years ago, induced pluripotent stem cells (iPSCs) have been used to model a multitude of “diseases in a dish” by utilizing lines derived from a relatively small number of diseased and healthy donors (Avior et al., 2016). Several recent initiatives, however, have begun to scale the generation of iPSC lines to create large banks of hundreds or thousands of iPSCs derived from diverse donors for studying stem cells and differentiated cell types in a variety of genetic backgrounds (McKernan and Watt, 2013; Panopoulos et al., In press; Streeter et al., 2017). Using these large banks of iPSCs for experiments requires an understanding of how donor genetic background affects various iPSC phenotypes. Genetic background has been shown to affect gene expression in iPSCs, but only recently have sufficiently large collections of iPSCs with corresponding genotype and gene expression data become available that enable genotype-expression association studies (Banovich et al., 2016; Kilpinen et al., 2016; Rouhani et al., 2014; Thomas et al., 2015). Understanding the effect of genetic background on iPSC gene expression is critical for estimating pluripotency and differentiation efficiency, studying gene dysregulation in disease, and comparing differentiated tissues to somatic tissues.
A common approach for investigating the effect of genetic background on gene expression is expression quantitative trait loci (eQTL) mapping. eQTLs are genomic regions that harbor genetic polymorphisms associated with the mRNA expression of a gene. Over the last 15 years, eQTL mapping has been performed in a variety of cell types and model organisms and has contributed to our understanding of how genetic variants regulate gene expression (Albert and Kruglyak, 2015). Prior eQTL studies in other cell types have focused on correlating single nucleotide polymorphisms (SNPs) and small insertions and deletions (indels) with gene expression, but several studies have also established the importance of copy number variant (CNV) eQTLs (Gamazon et al., 2011; Gamazon and Stranger, 2015; Handsaker et al., 2015; Stranger et al., 2007; Sudmant et al., 2015). Recent advances in high-depth whole genome sequencing (WGS) combined with new CNV-calling algorithms greatly enhance our ability to investigate how CNVs regulate gene expression (Chiang et al., 2016; Handsaker et al., 2015; Layer et al., 2014). Understanding the effect of inherited CNVs is particularly important for modeling complex phenotypes using iPSCs because CNVs are more likely to affect gene expression and be associated with complex traits than SNPs or indels (Sudmant et al., 2015).
Rare variants (minor allele<~0.5% in general population) constitute another class of variation whose effect on gene expression has been poorly assessed despite their established importance for disease (UK 10K Consortium, 2015). Some studies have leveraged unique family structures (Li et al., 2014) or deep targeted sequencing (Zhao et al., 2016) to investigate the effect of the vast number of rare regulatory variants on gene expression but only recently has it become feasible to use high-depth WGS to identify rare variants and quantify their effect on gene expression in a large set of subjects as explored in (Zeng et al., 2015) and recent preprints (Li et al., 2016; Pala et al., 2016). Thus the extent to which rare variants contribute to gene expression is not known, and it remains difficult to predict which of the estimated 40k-200k rare variants per genome may affect gene expression (1000 Genomes Project Consortium, 2015).
In this study, we leverage high-depth WGS to explore the genetic regulation of gene expression in a set of 215 iPSC lines. We demonstrate that iPSCs are well-powered for eQTL mapping and have a distinct regulatory landscape relative to somatic tissues. We functionally annotate the iPSC eQTLs and show they are enriched in stem cell regulatory elements and for overlapping the binding sites of transcription factors (including NANOG and POU5F1) important for establishing and maintaining pluripotency. To identify putative causal variants underlying the eQTL signals, we identify variants that are both associated with gene expression and alter transcription factor binding. We show that these putative causal variants are associated with allelic transcription factor binding in iPSCs and validate several examples in vivo. We observe that a large proportion of common CNVs associated with gene expression levels are located in intergenic regulatory regions. We also find that rare genic CNVs have relatively large effects on gene expression that can be positive or negative dependent on their location relative to the gene while rare promoter SNVs overall have a small negative effect on gene expression. Finally, we investigate X chromosome reactivation during reprogramming for iPSC lines from female donors and find that overall X reactivation is heterogeneous across lines but that the reactivation statuses of nearby genes are correlated. This work provides a stem cell-specific map of genetic regulators of gene expression that can be leveraged by future studies investigating the genetic basis of gene expression in stem cells and stem cell models of disease and development.
Results
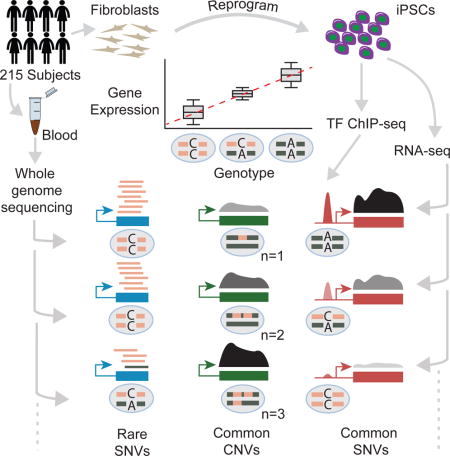
To investigate the genetic regulation of gene expression in iPSCs, we generated 30x germline WGS and RNA sequencing (RNA-seq) data for 215 human iPSC lines from a diverse set of donors (median age 48.3, 55% female) described in (Panopoulos et al., In press). The donors consist of both unrelated individuals as well as families and represent several ancestries although the majority (66%) are European. We used the high-depth WGS data to identify 22,461,624 single nucleotide variants (SNVs) and insertions/deletions (indels) using GATK and 15,735 CNVs using LUMPY and GenomeSTRiP after filtering for 1% minor allele frequency among our 215 subjects and violations of Hardy Weinberg equilibrium (Methods) (Handsaker et al., 2015; Layer et al., 2014; McKenna et al., 2010). We verified that the WGS samples were concordant with reported sex, family structures, and ethnicity and found an average of 99.9% concordance with genotypes from HumanCoreExome arrays demonstrating the quality of our variant calls.
eQTL Mapping in iPSCs
We used gene expression estimates from RNA-seq and germline variant calls to map eQTLs in 215 iPSC lines from different donors. We identified eQTLs using a permutation approach similar to (GTEx Consortium, 2015) but used EMMAX (Kang et al., 2010) to calculate association p-values that accounted for relatedness amongst our donors (Methods). Of the 17,805 autosomal genes tested we found 5,746 (32%) with eQTLs (eGenes) including 4,622 protein coding genes (Figure 1A, Table S1). The lead (most significant) variant was a SNV, indel, or CNV for 4,988, 1,376, and 108 eGenes respectively (some eGenes had multiple variants with equal significance) (Table S2). Consistent with previous eQTL studies, lead variants were enriched around the transcription start sites (TSSs) of genes (Figure S1), and 4.8% of eGenes had evidence of allele specific expression (ASE) compared to 1.7% of genes without eQTLs which supports the presence of cis eQTLs at these loci. We also found on average 93% agreement for lead SNV direction of effect compared to 44 GTEx v6 tissues demonstrating that our eQTLs are of high quality. As in previous studies, we observed an enrichment of lead eQTL variants among associations from genome-wide association studies (Table S1) (GTEx Consortium, 2015).
Figure 1.
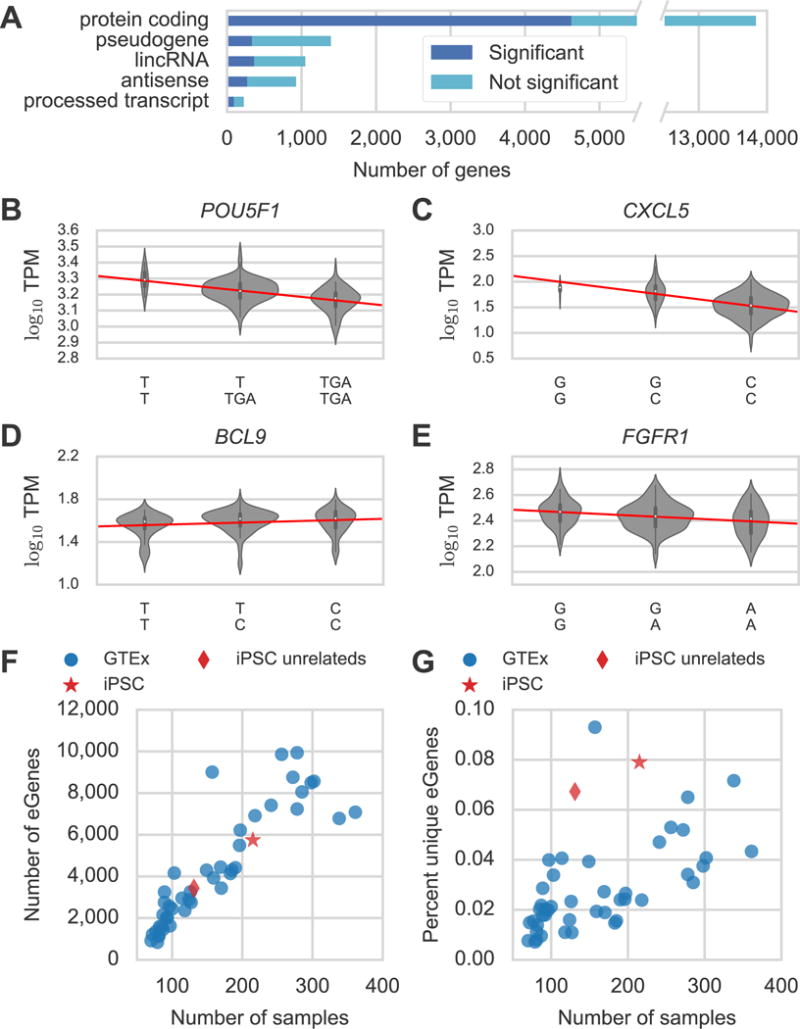
Summary of eQTL Results and Power Analysis. (A) Number of genes tested (green) and significant (blue) by Gencode gene type (see Table S1 for all gene types). (B-E) log10 RSEM TPM gene expression estimates stratified by lead variant genotype for (B) POU5F1, (C) CXCL5, (D) BCL9, and (E) FGFR1. The x-axis is labeled with the genotypes for the lead variant for each eQTL. We used residual expression values to identify eQTLs but plot raw TPM here to demonstrate the effect of the eQTL on the raw expression data. (F) Number of eGenes and (G) percent unique eGenes versus number of samples for 44 GTEx v6 tissues (blue circles), 131 unrelated subjects from this study (red diamond), or all 215 subjects from this study (red star). The outlier GTEx tissue in (G) is testis. See also Figure S1 and Tables S1 and S2.
Since gene expression is often used to estimate stem cell pluripotency, we compared our eGenes to nine stem cell marker genes from (Tsankov et al., 2015) and found that three (CXCL5, IDO1, and POU5F1) had eQTLs (Figure 1B–C, Table S2). The lead variants for these four genes explained respectively 19%, 11%, and 18% of the variance in gene expression in a model using only batch, sex, and donor age as covariates. We also identified eQTLs for 36 of 191 genes involved in stem cell population maintenance (GO:0019827) such as the oncogene BCL9 and the developmental regulator FGFR1 (Ashburner et al., 2000) (Figure 1D–E, Table S2) indicating genes relevant to pluripotency and differentiation also contain eQTLs.
To investigate the power to detect eQTLs in iPSCs, we compared the number of eGenes discovered in our study to the number identified in 44 GTEx v6 tissues, taking sample numbers in both studies into account (Figure 1F). Since GTEx uses unrelated subjects, we mapped eQTLs again using 131 of our 215 individuals who are genetically unrelated individuals and found eQTLs for 3,434 of 17,805 genes compared to 5,746 eGenes for all 215 subjects. The number of eGenes for both the 131 unrelateds and all 215 samples follow the same general trend observed in the GTEx data of an increase of about 30 eGenes per additional sample indicating that iPSCs are powered similarly to GTEx tissues for detecting eQTLs (Figure 1F). Since GTEx mostly focuses on somatic tissues, we hypothesized that the iPSCs might contain more unique eGenes (i.e. not found in other tissue types) than a typical GTEx tissue. To test this, we compared the percentage of eGenes that were unique to a given tissue relative to all GTEx eGenes plus the iPSC eGenes reported here (Figure 1G). GTEx tissues with more samples have a higher percentage of unique eGenes, with an increase of roughly 1.4% unique eGenes per 100 samples (excluding testis), likely reflecting the discovery of small effect size, tissue-specific eQTLs. Given this trend in the GTEx tissues, we would expect 2.4% (95% confidence interval [0.1%, 4.6%], excluding testis) of the 3,434 eGenes identified using the 131 iPSCs to be unique to iPSCs but instead observed that 6.8% of these eGenes are unique to iPSCs. Only testis (9.3% unique eQTLs) had a higher fraction of unique eQTLs consistent with testis as an outlier for gene expression and eQTLs (Mele et al., 2015). These results demonstrate that iPSCs are well-powered for identifying eQTLs and that the gene regulatory landscape of iPSCs differs significantly compared to the primary tissues and transformed cell lines in GTEx.
iPSC eQTLs Enriched in Stem Cell Regulatory Regions
To determine whether our eQTLs correspond to annotated stem cell regulatory regions, we investigated whether noncoding lead eQTL SNVs and indels were more likely to overlap stem cell regulatory regions compared to regulatory regions for other cell types. We calculated the enrichment of the 4,616 noncoding lead variants in DNase hypersensitivity sites (DHSs) from 53 Roadmap Epigenomics cell types by determining whether lead variants overlapped DHSs more often than nucleotides in 5kb windows centered on the lead variants (Figure 2A, Table S3, Methods) (GTEx Consortium, 2015; Roadmap Epigenomics Consortium, 2015). Although the lead eQTL variants are enriched in DHSs from most of the Roadmap cell types due to shared regulatory architecture across cell types, the enrichments are most significant in DHSs from hESCs and iPSCs consistent with these lead variants being located in stem cell regulatory regions. We also calculated the enrichment of noncoding lead SNVs and indels for 209 ENCODE DHS experiments comprising 134 different cell types and again found that noncoding lead variants enrichments were most significant in DHSs from stem cells followed by in vitro differentiated cells which likely reflects incomplete/heterogeneous differentiation or retention of some stem cell regulatory features in these lines (Figure 2B, Table S3) (Encode Project Consortium, 2012). The 209 ENCODE DHS experiments included nine skin fibroblast experiments that ranked from the 19th to the 206th most significant, so there does not appear to be a strong signal of epigenetic memory for the somatic cell type that affects iPSC gene expression (Table S3).
Figure 2.

eQTL Functional Annotation Enrichments. -log10 Fisher exact enrichment p-values for 4,616 eQTL lead SNVs/indels in (A) Roadmap Epigenomics DNase hypersensitivity sites (DHSs) and (B) ENCODE DHSs. The replicate H1 hESC DHS experiments in (B) were performed in different laboratories which may account for their different levels of enrichment. (C) Fisher exact odds ratios for ENCODE H1 hESC transcription factor CHiP-seq peaks. Color indicates whether the enrichment was significant which can vary due to the number of ChIP-seq peaks for each particular mark. See also Table S3.
The fact that lead variants are enriched in DHSs from both hESCs and iPSCs agrees with previous work showing that these two cell types have highly similar gene expression and epigenetic marks (Choi et al., 2015; Rouhani et al., 2014) and enables us to use the substantial amount of functional genomics data publicly available for the H1 hESC line to annotate our eQTLs. We calculated the enrichment of the 4,616 noncoding lead SNVs and indels among peaks from 49 ENCODE H1 hESC transcription factor (TF) ChIP-seq experiments and found that NANOG and POU5F1 were the first and third most-enriched for non-coding lead variants consistent with these factors’ known roles in reprogramming and pluripotency (Figure 2C, Table S3). These results suggest that genetic variation in the binding sites for TFs such as NANOG, BCL11A, NANOG, and JUN (AP1) are particularly important for regulating gene expression in stem cells.
Disruption of Transcription Factor Binding Sites by eQTL Variants
While a single eQTL typically contains multiple variants associated with the expression of the eGene due to linkage disequilibrium, generally only one variant is the functional, or causal variant, termed the expression quantitative trait nucleotide (eQTN). Given that functional genomics annotations such as TF ChIP-seq or DHS peaks can help identify candidate eQTNs and altered TF binding is thought to be one of the primary causes of eQTLs (Gaffney et al., 2012; Pai et al., 2015), we investigated how many eQTL SNVs and indels overlapped TF ChIP-seq peaks and disrupted motifs associated with those TFs. To identify putative eQTNs (peQTNs) that disrupt TF binding, we focused on 5,606 of the 5,746 eGenes that did not overlap a CNV eQTL and did not have an eQTL predicted to cause NMD since these eQTLs are less likely to be caused by altered TF binding. We overlapped the 191,871 eQTL SNVs and indels associated with the expression of these 5,606 eGenes with H1 hESC ChIP-seq peaks from 40 ENCODE TF ChIP-seq experiments and identified 3,140 variants that both overlapped a ChIP-seq peak and disrupted a motif associated with that TF in (Kheradpour and Kellis, 2014) (Table S4). Though we did not consider distance to the TSS when identifying peQTNs, 54% of the peQTNs were within 20kb of the nearest TSS for the associated eGene consistent with previous estimates of the distribution of eQTLs around the TSS (Figure S2A) (Wen et al., 2015). 90% of the peQTNs overlap a DHS present in at least one of the four Roadmap stem cell lines and 61% overlap a DHS present in all four lines (Figure 3A). peQTNs were also four times more likely to interact with the promoter of the associated eGene according to ChIA-PET interactions from naive hESCs (OR=4.0, p<10−18, Fisher exact test) (Ji et al., 2016). These observations suggest that the peQTNs are located in active stem cell regulatory regions.
Figure 3.

peQTN Characteristics. (A) Number of stem cell DHSs overlapped by eQTNs for four stem cell lines from Roadmap Epigenomics (H1, H9, iPS DF 6.9, iPS DF 19.11). (B) Number of peQTNs per eGene for 1,526 eGenes with at least one peQTN. (C) Putative eQTN for MED30 that overlaps a CEBPB ChIP-seq peak and disrupts a known CEBPB motif. Scatter plot shows -log10 association p-value from EMMAX for peQTN (purple point), other significant eQTL variants (green points) and variants not associated with MED30 expression (blue points). ENCODE H1 hESC CEBPB ChIP-seq peaks (blue rectangles) and chromHMM chromatin state predictions (multi-color track) are displayed. (D) CTCF ChIP-seq peak coverage (z-score of log10 counts) from five iPSC lines for peaks containing peQTNs predicted to disrupt CTCF binding and that had evidence of CTCF allelic bias in the ChIP-seq data. Counts are stratified by the genotype of the peQTN: 0, 1, and 2 for low, intermediate, and high predicted binding of CTCF, respectively. The peak coverage is significantly associated with the peQTN genotype (r=0.087, p=0.039). (E) The P1 region with the reference allele drives expression in 96% of embryos while the alternate allele leads to a complete loss of expression. (F and G) Image showing tailbud stage Ciona embryo electroporated with (F) Snail P1 (ref. allele) > GFP or (G) Snail P1 (alt. allele) > GFP. Expression can be seen in the tail muscle for the reference allele. Images were taken at the same exposure time to allow for direct comparison. See also Figure S2 and Tables S4 and S5.
In total, the 3,140 peQTNs we identified correspond to 1,526 of the 5,606 eGenes. 50% of these 1,526 eGenes have only one peQTN and 92% have five or less peQTNs indicating that most eGenes have few peQTNs (Figure 3B). A lead variant was a peQTN for 20% of the 1,526 genes though 61% of the genes had a peQTN with a p-value within one order of magnitude of their lead variants. eGenes with second, independent eQTLs were more likely to have their peQTN p-value differ by more than one order of magnitude from the lead variant (p<10−12, Fisher exact test) suggesting that the presence of multiple eQTLs (some of which we cannot detect at this sample size) may partially explain why only 20% of lead variants were identified as peQTNs. 60% of the 1,526 eGenes had a peQTN that disrupted a known motif for the overlapped TF ChIP-seq peak while the remaining 40% had a peQTN that disrupted a novel motif for the TF from (Kheradpour and Kellis, 2014). In some cases, these novel motifs may be similar to known motifs for other TFs which may be due to cooperative/interfering binding or motif similarity (Kheradpour and Kellis, 2014). Figure 3C shows an example of a peQTN for MED30, a component of the Mediator complex. The peQTN is a one base pair indel in the promoter of MED30 that overlaps a ChIP-seq peak for CEBPB in the H1 hESC line and disrupts a known motif for CEBPB. This indel is a strong candidate eQTN for the MED30 eQTL.
We next sought evidence that the peQTNs we identified cause differential TF binding. (Maurano et al., 2015) tested ~360k heterozygous SNVs located in DHSs for allelic bias caused by differential TF binding in vivo and found that 18% of tested variants affected TF binding. Of the 191,871 eQTL variants we used to identify peQTNs, (Maurano et al., 2015) assayed 13,664 including 992 peQTNs. We found that 38% of the 992 peQTNs showed evidence for altered TF binding in (Maurano et al., 2015) compared to only 19% of the 12,672 eQTL variants assayed by (Maurano et al., 2015) that we did not classify as peQTNs. Thus peQTNs are highly enriched for altering TF binding relative to eQTL variants that we did not classify as peQTNs (OR=2.5, p<10−37, Fisher exact test) and relative to all ~360k variants tested by Maurano (OR=2.8, p<10−47, Fisher exact test). We also performed CTCF ChIP-seq for iPSCs from five subjects to test whether peQTNs that were predicted to disrupt CTCF binding showed evidence of allelic CTCF binding. We tested 73 heterozygous peQTNs on average in each sample and found that 207 of the 366 (57%) peQTNs tested had significant allelic bias in CTCF binding (binomial, p<0.005). We also found the number of reads per CTCF peak was significantly associated with predicted CTCF binding affinity for peQTNs with significant allelic bias (r=0.087, p=0.039, Figure 3D). These results indicate that many peQTNs disrupt TF binding and provide further evidence that the peQTNs we have identified are good candidate eQTNs.
To provide further validation of our peQTNs, we selected nine enhancer regions (E1–E9) and one promoter region (P1) containing peQTNs to test for function in vivo in the urochordate Ciona intestinalis, a member of the sister group to the vertebrates (Delsuc et al., 2006). Ciona is an excellent system in which to screen for the impact of sequence variation on regulatory function as many of the transcriptional programs used during development are evolutionarily conserved with vertebrates (Abitua et al., 2015; Farley et al., 2015; Stolfi et al., 2015). For the enhancer regions, constructs containing either the reference or alternate allele attached to a minimal super core promoter (Juven-Gershon et al., 2006) and GFP were electroporated into Ciona fertilized eggs; for the promoter region, the tail muscle enhancer Snail (Erives et al., 1998) was included upstream. 6/10 regions tested drove expression in Ciona embryos and 4/6 of these showed differential expression between the reference and alternate alleles (p<0.05, Fisher exact test, Figures 3E, S2B,C, Table S5). The allele with higher expression agreed with the iPSC eQTL for 3/4 regions with differential expression. The most striking result was seen for the promoter region where the reference allele drove expression in 96% of embryos while the alternate variant lead to a complete loss of expression (Figure 3E–G). The peQTN for P1 disrupts a TATA motif, overlaps a TBP ChIP-seq peak, and is near a TAF1 peak in the H1 line. However, the alternate allele also creates a binding site for the zinc finger transcriptional repressor SLUG that is upregulated following the addition of reprogramming factors (Liu et al., 2013). The presence of a SLUG binding site is consistent with evidence showing that SLUG represses transcription by preventing RNA Pol II and associated proteins from binding to the promoter region (Chiang and Ayyanathan, 2013). Overall these results suggest that our peQTNs provide a good starting point for identifying functional regulatory variants that cause eQTLs.
Copy Number Variant eQTLs
To examine the effect of CNVs on gene expression, we used our high-depth WGS to identify 15,281 autosomal biallelic CNVs that were within 1Mb of at least one TSS and included these CNVs when testing for eQTLs as described above. We found significant CNV-expression associations (CNV eQTLs) for 247 genes including 108 genes for which the CNV was the lead variant (47 deletions, 41 duplications, and 28 mixed duplications/deletions) and 64 genes whose expression was associated with a CNV but not a SNV or indel. Lead CNVs had larger effect sizes than lead SNVs and indels (p<10−9, Mann Whitney U, Figure 4A), and we observed a positive correlation between copy number and gene expression for 82% of lead CNVs (Figure S3A). Consistent with previous reports, a large fraction (59%) of CNV eQTLs did not overlap their associated eGenes and therefore may regulate their associated eGene in trans (Gamazon and Stranger, 2015; Stranger et al., 2007; Sudmant et al., 2015). We found that these intergenic CNV eQTLs are generally positively correlated with gene expression (p<10−4, binomial test) and are enriched for marks of active regulatory regions but not repressive marks or marks of active transcription relative to CNVs that were not eQTLs (Figure 4B,C) likely because the CNV eQTLs are longer (p<10−74, Mann Whitney U, median 2,386 bp versus 528 bp) and closer to transcription start sites (p<10−75, Mann Whitney U, median 3,547 bp versus 12,390 bp) than CNVs that were not eQTLs (Figure S3B,C). These data suggest that intergenic CNV eQTLs can affect gene expression levels by altering the dosage of intergenic regulatory regions.
Figure 4.

CNV eQTL Effect Sizes and Functional Annotation. (A) Density plot of absolute effect size for eQTLs with or without CNV lead variants. (B) Effect sizes for lead CNVs for eGenes where no significant CNV overlaps the eGene. (C) Enrichment p values (Fisher exact test) of Roadmap stem cell DHS and histone modification ChIP-seq peaks in lead CNVs for eGenes where no significant CNV overlaps the eGene. Different points for each mark represent different Roadmap stem cell lines. (D) Gene expression estimates for seven genes associated with a single mCNV in 131 unrelated donors. (E) Genomic location of mCNV on chromosome seven along with six of seven associated genes (indicated by boxes). The mCNV overlaps a CEBPB ChIP-seq peak, DHS, and predicted enhancer from the H1 hESC line.
It was recently reported that multiallelic CNVs (mCNVs) are an important class of CNVs that can affect gene expression (Handsaker et al., 2015). Since EMMAX is limited to testing for associations with biallelic variants and cannot test for associations with multiallelic loci, we identified mCNV eQTLs by regressing gene expression estimates against genotype using a linear model for the 131 unrelated individuals. After filtering (Methods), we identified 152 mCNVs segregating in the 131 unrelated individuals that were within 1 Mb of one or more genes and found mCNV eQTLs for 90 genes of which 22 overlapped an associated mCNV and 68 did not. The effect sizes for mCNV eQTLs were again skewed toward positive associations between gene expression and copy number for both mCNV eQTLs that overlapped genes and those that did not (Figure S3D,E) indicating that mCNVs may also affect gene expression by altering the dosage of regulatory regions. For example, we identified a 2kb mCNV on chromosome seven whose diploid copy number estimates ranged from one to eight and that was associated with the expression of seven nearby genes (Figure 4D). While this mCNV slightly overlaps one of the genes it is associated with, it also overlaps a DHS, CEBPB TF ChIP-seq peak, and predicted enhancer in the H1 hESC line suggesting that the CNV alters gene expression in the region by changing the copy number of this regulatory region (Figure 4E). Although most of the intergenic mCNV eQTLs were associated with the expression of only one or two genes, the bias toward positive associations between copy number and gene expression and the scaling of gene expression with the dosage of intergenic regions indicates that intergenic CNVs can cause eQTLs.
Effect of Rare Variants on Gene Expression
Rare variants are another class of variants whose effect on gene expression has been difficult to investigate because accurate identification of rare variants within an individual requires high-depth WGS, and large sample sizes are needed to achieve sufficient statistical power to detect rare variant associations. While we expect to observe many rare variants across our 215 subjects, most of these will not fall in genes or regulatory regions and are not likely to affect gene expression. Therefore, to investigate the effects of noncoding rare variants on gene expression, we decided to focus on rare variants located in the promoters of expressed genes. We identified 65,530 SNVs that (1) were located in the promoters of 17,820 robustly expressed autosomal genes, (2) overlapped a DHS from at least one of the four Roadmap stem cell lines (Figure 2A), (3) had only one minor allele observed among the 131 unrelated subjects, and (4) were either not observed in 1000 Genomes or whose minor allele frequency was less than 0.5% in all 1000 Genomes populations (1000 Genomes Project Consortium, 2015). We refer to these 65,530 SNVs as rare promoter DHS SNVs (rpdSNVs). In total, 14,589 of 17,820 robustly expressed genes had an rpdSNV in at least one of the 131 unrelated subjects.
To determine the effect of rpdSNVs on gene expression, we stratified gene expression estimates based on the presence of an rpdSNV (Methods) and found that expression estimates for samples with rpdSNVs were slightly lower than estimates for samples without rpdSNVs indicating that the presence of an rpdSNV has a small but significant effect on gene expression (p=0.00088, Mann Whitney U, Figure 5A). Additionally, genes were more likely to have significant ASE in samples with an rpdSNV versus samples without an rpdSNV (OR=1.23, p<10−8, Fisher exact test) consistent with rare variants affecting gene expression in cis. It was reported previously that evolutionary constraint and functional annotations can help predict which rare variants may affect gene expression (Li et al., 2014) so we filtered the rpdSNVs according to phyloP conservation and CADD scores (Kircher et al., 2014; Pollard et al., 2010). We found that the bias toward lower expression estimates was stronger for genes with an rpdSNV with a CADD Phred score greater than 20 (p<10−4, Mann Whitney U) or a phyloP score greater than 3 (p<10−4, Mann Whitney U, Figure 5B). We also observed higher rates of ASE among genes with rpdSNVs with a CADD Phred score greater than 20 (OR=1.66, p=0.0008, Fisher exact test) or a phyloP score greater than 3 (OR=1.69, p=0.0002, Fisher exact test) compared to genes that did not have an rpdSNV. These results show that rpdSNVs that affect gene expression generally cause a decrease in expression and that rpdSNVs are more likely to affect gene expression if they are in conserved sequences or have higher CADD scores.
Figure 5.

Effect of Rare Variants on Gene Expression. Distribution of gene expression estimates (PEER residual z-scores, Methods) for genes (A) with (green) or without (blue) a rare promoter DHS SNV (rpdSNV) and (B) without an rpdSNV (blue), with an rpdSNV with CADD Phred greater than 20 (green), or with an rpdSNV with a phyloP score greater than three (orange). Distribution of gene expression estimates for genes (C) without rare genic duplications (blue), with rare genic duplications (green), or with rare exonic duplications (orange) and (D) without rare genic deletions (blue), with rare genic deletions (green), or with rare exonic deletions (orange). See also Figure S3.
We next asked whether rare CNVs that overlap genes may affect gene expression by altering the dosage or structure of the overlapped gene. We defined rare genic CNVs as CNVs that overlapped introns and/or exons of genes and were observed in only one of the 131 unrelated subjects in our study. In total, we identified 428 rare genic duplications and 2,122 rare genic deletions. We stratified expression estimates into three groups based on the presence or absence of either a rare genic duplication or deletion for a given gene and subject. We found that the 428 rare genic duplications had a much stronger effect on gene expression than rpdSNVs and generally caused increased gene expression (Figure 5C). This effect was stronger if we restricted to the 224 rare duplications that were predicted to overlap exons as opposed to the larger set of deletions which includes some deletions that are only intronic (p=0.015, Mann Whitney U). As observed for rpdSNVs, genes were much more likely to have significant ASE in subjects with rare genic duplications (OR=6.26, p<10−10, Fisher exact test) with nearly 16% of such genes demonstrating significant ASE. The presence of higher rates of ASE among genes with rare duplications indicates that the altered expression of these genes is likely caused by these duplications. As opposed to duplications, we found that the 2,122 rare genic deletions generally caused lower expression (Figure 5D). This effect was much stronger for the 507 rare deletions that were predicted to overlap exons (p<10−12, Manny Whitney U). 9.1% of genes with rare exonic deletions in a given sample had significant ASE compared to 2.9% of the genes that did not have rare exonic deletions (OR=3.39, p=0.0002, Fisher exact test). These results indicate that rare genic CNVs are more likely to affect gene expression than rpdSNVs and generally have larger effects whose direction is dependent on the CNV type.
X Reactivation Status Varies According to Gene Chromosomal Position
X inactivation (Lyon, 1961) has been studied in iPSCs derived from female donors to determine the behavior of the inactive X chromosome during reprogramming and passaging (Lessing et al., 2013; Pasque and Plath, 2015) but the heterogeneity of X chromosome reactivation (XCR) across a large set of systematically reprogrammed lines is unknown. Since our iPSCs are clonally derived from single fibroblasts, female-derived iPSCs should have one inactive and one active X unless the inactive X has been reactivated during reprogramming or passaging. iPSCs with residual X inactivation should have a higher amount of ASE for genes on the X chromosome relative to autosomal genes (i.e. ASE for genes on the X chromosome is a proxy for X inactivation). We calculated the percentage of X chromosome and autosomal genes with significant ASE per sample for 144 RNA-seq samples from the 116 iPSC lines derived from female donors (predominantly assayed at passage 12) and found that the X chromosome is highly enriched for ASE relative to autosomes with an average of 44% of X chromosome genes displaying significant ASE per sample compared to only 3% of autosomal genes per sample. We identified 120 robustly expressed X chromosome genes and stratified each gene’s expression estimates into two groups based on whether or not the gene had significant ASE in a given sample. We calculated the average expression of each gene in the two groups and observed that 78% of the genes had lower average expression in the group of samples with significant ASE consistent with allelic silencing of these genes by X inactivation (Figure 6A). These results indicate that X inactivation persists at some level for most iPSCs derived from female subjects and affects the gene expression of X chromosome genes.
Figure 6.
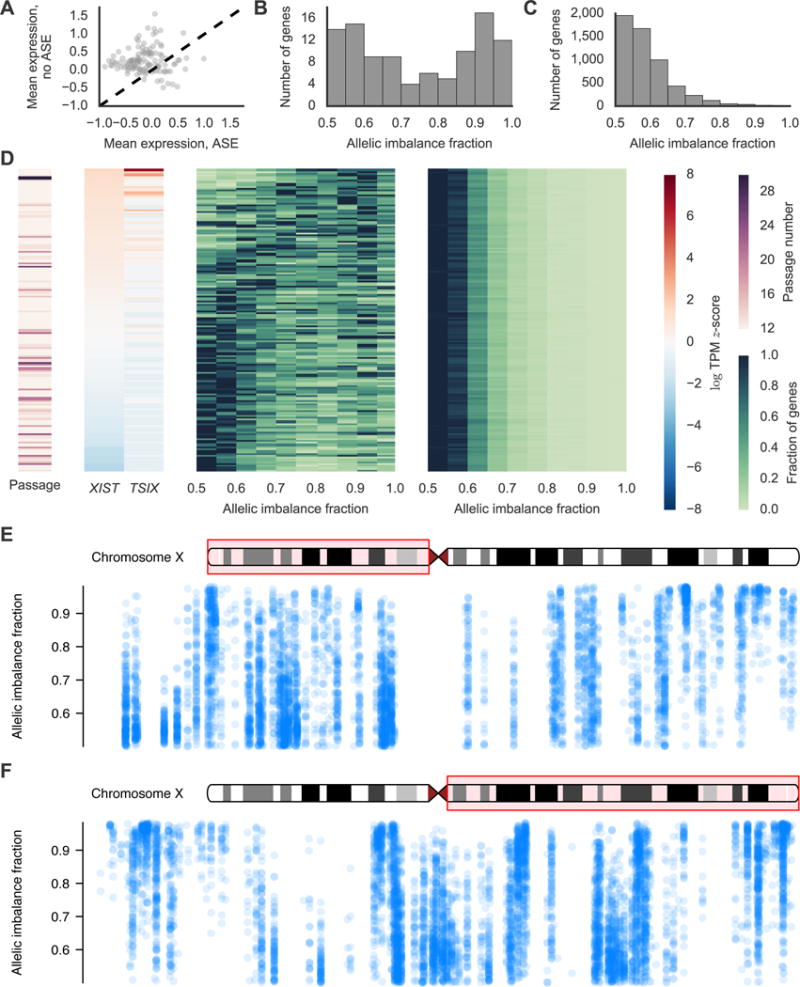
Heterogeneity of X Chromosome Reactivation Following Reprogramming. (A) Average expression for 120 X chromosome genes in samples with significant ASE versus samples without significant ASE. (B and C) Distribution of estimated allelic imbalance fractions (AIFs) for (B) X chromosome and (C) autosomal genes for one representative RNA-seq sample. AIF is the percentage of transcripts estimated to come from the haplotype that is more expressed. (D) From left to right, the heatmaps show passage; XIST and TSIX expression; X chromosome AIF distribution; and autosomal AIF distribution for each RNA-seq sample from female-derived iPSCs. Each row corresponds to one one sample. Samples were sorted by XIST expression before plotting. (E and F) Estimated AIFs across the (E) p and (F) q arms of the X chromosome. Each point represents an estimate of the AIF for a gene/sample pair. Box 1 shows a largely reactivated cluster of genes with most AIFs near 50% while box 2 shows a cluster of genes that have not been reactivated with AIFs closer to 100%.
To examine the heterogeneity of XCR across the iPSCs, we defined the strength of ASE for a given gene as the percentage of RNA transcripts estimated to originate from the parental haplotype with higher expression, referred to as the allelic imbalance fraction (AIF) (Mayba et al., 2014). The distribution of AIFs for X chromosome genes was bimodal with some genes showing relatively balanced expression (AIF near 0.5) and other genes displaying nearly mono-allelic expression (AIF near 1.0) consistent with some X chromosome genes remaining silenced following reprogramming (Figure 6B). In contrast, the AIFs for most autosomal genes was near 0.5 with few genes showing evidence for strong allelic bias (Figure 6C). Stratifying the AIFs by sample showed that there is considerable variation between samples with some iPSC displaying low levels of XCR and others displaying high levels of XCR (Figure 6D). The percentage of X chromosome genes with significant ASE per sample (a proxy for the overall amount of XCR per sample) is correlated with XIST (r=0.72, p<10−24, Spearman) gene expression consistent with previous reports that XIST is down-regulated as the inactive X is reactivated (Pasque and Plath, 2015). We also found that TSIX expression was positively correlated with the percentage of X chromosome genes with significant ASE (r=0.51 and p<10−11, Spearman). XIST (r=−0.18, p=0.029, Spearman) and TSIX (r=−0.17, p=0.044, Spearman) expression are also negatively correlated with passage although passage is not correlated with the percentage of X genes with significant ASE (r=−0.07, p=0.43). However, most of our iPSC lines were at passage 12 so it is possible that we are not powered to find this latter association. These results suggest that XIST and TSIX are downregulated as the inactive X is reactivated during early passages.
While we observed that the overall amount of XCR differs between lines (Figure 6C), we also asked whether the reactivation status of genes was correlated with respect to their location on the X chromosome. We plotted the AIF estimates for each gene in each sample versus the position of the gene on the X chromosome and observed that clusters of nearby genes tended to show similar levels of reactivation even in different lines (Figure 6E,F). Our data suggest that while the overall amount of XCR differs between lines, reactivation follows the same physical pattern in different lines with some clusters of nearby genes consistently becoming reactivated faster than others.
Discussion
We present here a map of the genetic determinants of gene expression in stem cells derived using WGS and RNA-seq from 215 systematically reprogrammed human iPSCs. Large sets of iPSCs are a promising system for exploring the genetics of complex traits and diseases (McKernan and Watt, 2013; Pai et al., 2015). This work underscores the suitability of iPSCs for genetic association studies and contributes to our understanding of how genetic variation affects gene expression in stem cells. These stem cell eQTLs will be useful for dissecting the regulatory architecture of gene expression in both normal stem cells and disease models.
Our results show that iPSCs are powered similarly to GTEx tissues for identifying genetic variants associated with gene expression (eQTLs) and have more unique eQTLs than expected compared to GTEx tissues suggesting that iPSCs have a distinct gene regulatory landscape. We based our eQTL mapping strategy on the GTEx methodology to enable this comparison, though it is possible technical differences between the two studies could contribute to the increased amount of unique iPSC eQTLs. In total we found 5,746 genes with eQTLs including 39 genes involved in stem cell population maintenance. iPSC eQTLs may affect gene expression-based approaches for estimating pluripotency and differentiation efficiency though it is unclear whether the variation in gene expression caused by eQTLs outweighs expression variation due to environmental factors. It may be necessary to perform differentiation assays for hundreds or thousands of lines to identify genetic variation that affects iPSC pluripotency.
Since altered TF binding has been proposed to be one of the primary causes of eQTLs (Pai et al., 2015), we identified putative causal variants (peQTNs) that overlap H1 hESC TF ChIP-seq peaks and disrupt TF motifs. We used ChIP-seq for CTCF to demonstrate that many peQTNs predicted to disrupt CTCF binding are associated with allelic CTCF binding in vivo. We also demonstrated that several peQTNs have differential expression between the reference and alternate alleles in Ciona intestinalis embryos. We were able to identify peQTNs for 27% of eGenes indicating that it may be necessary to profile more TFs or explore other mechanisms in order to dissect the majority of eQTLs. Interestingly, the lead variant was identified as the peQTN for only 20% of the 1,526 eGenes although the peQTN p-value was within one order of magnitude of the lead for 61% of the genes. The association p-values can be affected by a number of factors including the presence of multiple independent eQTLs. These results may be improved by utilizing methods for jointly identifying multiple eQTLs and fine mapping causal variants (Veyrieras et al., 2008; Wen et al., 2015).
We found that rare SNVs in promoters can affect gene expression and that this effect is stronger for conserved variants or those that overlap functional annotations, consistent with previous reports (Li et al., 2014; Zhao et al., 2016). Our results suggest that these rare variants typically act to decrease gene expression. We also found that though there are less rare genic CNVs than rare promoter SNVs, they are more likely to affect expression. It is notable that somatic variants that arise during the reprogramming process may have similar effects as inherited rare variants, and we anticipate future studies may benefit from genotyping iPSCs and germ cells to profile somatic variants genome-wide and incorporate them into association analyses.
We investigated the heterogeneity of XCR following reprogramming from female donors and found that most samples retain some amount of silencing on the X chromosome although the amount differs from sample to sample. We found that all lines share similar physical reactivation patterns across the X chromosome, with clusters of genes in some areas escaping silencing more quickly than clusters of genes in other areas. It has been suggested that human iPSCs do not undergo XCR and that increased expression of X-linked genes is due to instable X inactivation during iPSC passaging (Pasque and Plath, 2015; Tchieu et al., 2010). Given that most of our iPSCs are only at passage 12, our results suggest that XCR does occur in human iPSCs though it is not complete. It may be the case that XCR will complete upon further passaging or regress back to an inactive X. The differing rates of reactivation between samples and across the X chromosome shown here will need to be accounted for when investigating X-linked molecular quantitative traits, modeling diseases, or using iPSCs for therapeutics.
iPSCs are a promising system for mapping expression and other molecular trait QTLs for several reasons including their ability to self-renew and differentiate into other cell types (Pai et al., 2015). Genetic association analyses in iPSCs and differentiated cell types are not limited to gene expression or other molecular phenotypes like methylation levels but can also be extended to physiological phenotypes like electrophysiological responses or cellular phenotypes like cell survival after drug treatment (Avior et al., 2016). Merging “disease in a dish” modeling approaches with large-scale genetic association analyses like the one presented here will be useful for dissecting complex diseases and drug-genotype interactions and will likely become an important strategy for exploring the genetic and molecular causes of disease.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat Polyclonal Anti-CTCF | Santa Cruz Biotechnology | sc-15914 X; RRID:AB_2086899 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| AllPrep RNasy Blood & Tissue Kit | Qiagen | Cat no: 80204 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat no: 69506 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat no: RS-122-2103 |
| Deposited Data | ||
| Sequencing data except whole genome sequencing data | This paper | phs000924 |
| Whole genome sequencing data | This paper | phs001325 |
| Code | This paper | https://github.com/frazer-lab/cardips-ipsc-eqtl |
| Gencode Genes | Harrow et al., 2012 | https://www.gencodegenes.org/ |
| ENCODE | https://www.encodeproject.org/ | |
| Roadmap Epigenomics | Roadmap Epigenomics Consortium, 2015 | http://www.roadmapepigenomics.org/ |
| DNase hypersensitivity allelic bias data | Maurano et al., 2015 | NA |
| ChIA-PET data | Ji et al., 2016 | NA |
| CADD scores | Kircher et al., 2014 | http://cadd.gs.washington.edu/ |
| phyloP scores | Pollard et al., 2010 | http://hgdownload.cse.ucsc.edu/goldenpath/hg19/phyloP100way/ |
| GTEx | GTEx Consortium, 2015 | http://www.gtexportal.org/ |
| GO terms | Ashburner et al., 2000 | http://www.geneontology.org/ |
| Experimental Models: Cell Lines | ||
| iPSCORE human iPSC lines | WiCell | NA |
| Experimental Models: Organisms/Strains | ||
| Ciona Intestinalis type A | Wild population from San Diego Bay, collected by Scripps Institute of Oceanography and by MREP San Diego | NA |
| Recombinant DNA | ||
| SCP>uncGFP vector | Farley/Levine lab | NA |
| Sequence-Based Reagents | ||
| More than 10 therefore see Table S5 | ||
| Software and Algorithms | ||
| EMMAX | Kang et al., 2010 | http://csg-old.sph.umich.edu/kang/epacts/download/EPACTS-3.2.6.tar.gz |
| MBASED | Mayba et al., 2014 | https://www.bioconductor.org/packages/release/bioc/html/MBASED.html |
| PEER | Stegle et al., 2010 | https://github.com/PMBio/peer |
| SpeedSeq | Chiang et al., 2015 | https://github.com/hall-lab/speedseq |
| LUMPY | Layer et al., 2014 | https://github.com/arq5x/lumpy-sv |
| Genome STRiP | Handsaker et al., 2015 | http://software.broadinstitute.org/software/genomestrip/ |
| FASTQC | NA | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| GATK | McKenna et al., 2010 | https://software.broadinstitute.org/gatk/ |
| BWA | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/ |
| pyencodetools | NA | https://github.com/cdeboever3/pyencodetools |
| Sambamba | https://lomereiter.github.io/sambamba/ | |
| Biobambam2 | Tischler and Leonard, 2014. | https://github.com/gt1/biobambam2 |
| Other | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests should be directed to the corresponding author Kelly Frazer ([email protected]). For requests related to the Ciona experiments contact Emma Farley ([email protected]).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Sample collection and reprogramming
Skin biopsies were collected from 278 individuals and recorded sex, age, medical history, ethnicity, and relatedness were obtained through a questionnaire at enrollment. We successfully reprogrammed fibroblasts from 222 donors using Sendai virus. Pluripotency was assessed by measuring gene expression for pluripotency and mesoderm markers by RNA-seq and and by flow cytometry (>95% positive staining for keratan sulfate antigen Tra-1-81 and glycolipid antigen SSEA4) for a subset of lines (Panopoulos et al., In press). The Institutional Review Boards of the University of California at San Diego and of The Salk Institute approved the study and all subjects gave informed consent (Project #110776ZF).
Ciona intestinalis collection
For electroporation of regulatory regions into Ciona, wild-caught Ciona intestinalis (species type A) were collected in the San Diego bay by Scripps Institute of Oceanography staff or M-REP (San Diego, California).
METHOD DETAILS
RNA library preparation and sequencing
Total RNA was extracted from 222 iPSC lines using AllPrep DNA/RNA Mini Kit (Qiagen) following the manufacturer’s protocol. RNA quality was assessed based on RNA integrity number (RIN) using an Agilent Bioanalyzer. Any samples with RIN less than 7.5 were re-isolated. Libraries were prepared using the Illumina TruSeq stranded mRNA kit and sequenced using an Illumina HiSeq2500 (~11 samples per lane). Samples were sequenced to an average of ~22 million read pairs. Biological replicates were sequenced for some lines.
DNA library preparation and sequencing
Genomic DNA was isolated from blood (or in 19 cases directly from the fibroblasts, DNEasy Blood & Tissue Kit), quantified, normalized, and sheared with a Covaris LE220 instrument. The samples were normalized to 1 ug and submitted for whole genome sequencing. DNA libraries were prepared (TruSeq Nano DNA HT kit, Illumina), characterized in regards to size (LabChip DX Touch, Perkin Elmer) and concentration (Quant-iT, Life Technologies), normalized to 2–3.5nM, combined into 6-sample pools, clustered and sequenced on the HiSeqX (150 base paired-end). In total, germline whole genome sequencing (WGS) was performed for 274 subjects though only 222 were reprogrammed into iPSCs. WGS was performed at Human Longevity, Inc. (HLI).
ChIP library preparation and sequencing
Cells were cross linked with 1% formaldehyde for 10min at room temperature. For each sample 3×10^6 cells were lysed and sonicated using Covaris M220 for 10min. Sonicated chromatin were immunoprecipitated with anti-CTCF antibody (Santa Cruz Biotechnology, sc-15914 X). Libraries were prepared according the method described in (Panopoulos et al., In press; Yan et al., 2013) using Illumina TruSeq adapters, size selected for 300bp-500bp, and sequenced on Illumina HiSeq4000 for paired-end sequencing to obtain 100bp reads. Samples were sequenced to an average of ~44 million read pairs.
Ciona experiments
Electroporation
Wild caught adult C. intestinalis (species type A) were maintained in filtered seawater at 18°C, under constant illumination. Dechorionation, in vitro fertilization, and electroporation were carried out as described in (Christiaen et al., 2009). For each electroporation, typically, eggs and sperm were collected from 15 adults, 70 μg DNA was resuspended in 100 μL water. Embryos were fixed at the appropriate developmental stage for 15 min in 4% (wt/vol) formaldehyde. The tissue was then cleared in a series of washes of 0.01% Triton-X in PBS. Samples were mounted in 50% (vol/vol) glycerol in PBS with 2% (wt/vol) DABCO compound for microscopy. Differential interference-contrast microscopy was used to obtain transmitted light micrographs with a Zeiss Axio Imager A2, using the ×20 EC Plan Neofluar objective. The same microscope was used to obtain GFP images. All constructs were electroporated at least twice in two completely separate experiments (biological replicates). For experiments testing enhancer regions embryos were fixed at larval stage, for experiments to test the promoter embryos were fixed at late tailbud stage.
Acquisition of images
For enhancers that were being compared, images were taken on the same day and from electroporations performed on the same day, using identical settings. For images, embryos were chosen that represented the average from counting data. Images are rotated and cropped, but have no other manipulations. In each figure, the same exposure time for each image is shown to allow direct comparison.
Isolation of regulatory regions for reference and alternate alleles
Regulatory regions were amplified using Phusion High Fidelity Master mix (NEB M0531S) following their protocol. The genomic DNA used for amplification was isolated from iPSC line 1_14 using DNeasy Blood & Tissue Kit, Qiagen; primers and their coordinate are listed in Table S5. The size of region amplified was dependent on the location of the regulatory region. These regions were cloned into a SCP>GFP vector. The SCP promoter was given to us by J. Kadonaga (UCSD) (Juven-Gershon et al., 2006). For the promoter region this was cloned downstream of a 504bp Snail enhancer isolated by (Erives et al., 1998).
Mutagenesis of regulatory region
For site directed mutagenesis, 100ng of the reference vector was used as a template in the PCR along with 2.5uM forward and reverse primer (Table S5). We used Phusion high Fidelity Master mix (NEB M0531S) for the PCR following their protocol. Following PCR, 2ul of DpnI (NEB R0176S) was added to the PCR reaction and incubated at 37°C for 1hr. 5ul of this product was transformed into chemically competent cells. Clones were then sequenced. Correct clones were grown up and midi prepped using NucleoBond® Xtra Midiprep kit (Macherey-Nagel, 740410.10).
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA sequencing analysis
Alignment and quality control
2×100 bp RNA-seq reads were aligned with STAR (2.5.0a) to the hg19 reference (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/hg19.2bit) using Gencode v19 splice junctions with default alignment parameters except –outFilterMultimapNmax 20, –outFilterMismatchNmax 999, –alignIntronMin 20, –alignIntronMax 1000000, –alignMatesGapMax 1000000 (Dobin et al., 2013; Harrow et al., 2012). Bam files were coordinate sorted using Sambamba (0.5.9) (Tarasov et al., 2015) and duplicate reads were marked using biobambam2 (2.0.21) bammarkduplicates (Tischler and Leonard, 2014).
We repeated library preparation and sequencing for samples that were outliers for percent uniquely mapped reads as reported by STAR or percent duplicate reads or 5′/3′ bias as estimated by Picard Tools. We identified outliers separately for each flow cell by converting each metric to a z-score; any z-scores with magnitude greater than 1.96 were considered outliers. Seven of the 222 samples that had outlying metrics after the second sequencing run were not used resulting in RNA-seq for 215 of the 222 lines. The minimum uniquely mapped read percentage was 86% and the median was 91%. The median percent duplicates was 16% and the maximum was 24%.
Gene expression
We estimated transcript and gene expression using the STAR transcriptome bam file and RSEM (1.2.20) rsem-calculate-expression (–seed 3272015 –estimate-rspd –forward-prob 0) (Li and Dewey, 2011).
Allele specific expression
Uniquely mapped reads that were not marked as duplicates were tested for mapping bias using the WASP mapping pipeline (van de Geijn et al., 2015). Reads that mapped uniquely to the same location after swapping in alternate alleles were used to calculate the coverage of heterozygous variants overlapping Gencode v19 exons for all exonic regions unique to one gene using the ASEReadCounter (-overlap COUNT_FRAGMENTS_REQUIRE_SAME_BASE, -U ALLOW_N_CIGAR_READS) from GATK (3.4–46) (Van der Auwera et al., 2013). MBASED was used to estimate per-gene and per-heterozygous variant allele specific expression (ASE) p-values (Mayba et al., 2014). Heterozygous variants that met the following criteria were used as input for MBASED: (1) coverage greater than or equal to 8, (2) reference allele frequency between 2–98%, (3) located in unique mappability regions according to wgEncodeCrgMapabilityAlign100mer track, (4) not located within 10 bp of another variant in a particular subject (heterozygous or homozygous alternate). Additionally, for heterozygous variants within 300 bp of each other, only one variant was used to avoid double counting variant coverage from the same read pair. These filters are based on the GTEx and MBASED ASE pipelines (GTEx Consortium, 2015; Lappalainen et al., 2013; Mayba et al., 2014). A gene was considered significant for ASE if the MBASED “p_val_ase” was less than or equal to 0.005 (GTEx Consortium, 2015).
DNA sequencing analysis
Alignment and quality control
We esitimated the quality of fastq files using FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were aligned against human genome b37 with decoy sequences (1000 Genomes Project Consortium, 2015) using BWA-mem and default parameters (Li and Durbin, 2009). The resulting bam files were sorted using Sambamba (Tarasov et al., 2015) and duplicate reads were marked using biobambam2 (Tischler and Leonard, 2014).
SNV/indel calling
The bam files were split into individual chromosomes to maximize the efficiency of the variant calling process on our cluster. We applied the GATK (McKenna et al., 2010) best-practices pipeline for variant calling that includes indel-realignment, base-recalibration, genotyping using HaplotypeCaller, and finally joint genotyping using GenotypeGVCFs (DePristo et al., 2011; Van der Auwera et al., 2013). We performed quality control for the genotypes of single nucleotide variants and indels using GATK’s Variant Quality Score Recalibration (VQSR) (Van der Auwera et al., 2013). We performed variant calling for sex chromosomes in males and females separately and resolved the pseudoautosomal regions of the sex chromosomes independently: pseudoautosomal regions of male X chromosome (chrX: 60001-2699520, chrX: 154931044 -155260560) were treated as diploid, whereas the rest of male X chromosome as well as chromosome Y were treated as haploid.
A series of quality control processes was performed to ensure the sample identity and sequencing quality prior to further analyses: 1) We estimated sex based on the heterozygosity rate on chromosome X; 2) Genetic relatedness among individuals was determined using identical by descent (IBD) and compared with the reported family structure; 3) Ethnicity was assessed using PCA along with samples from 1000 Genomes to check the consistency of self-reported race and estimated continental ancestry. We identified one sample for which the above analyses results were not consistent with the reported information and removed this sample from further analyses. 4) All the samples have been previously genotyped on Illumina HumanCoreExome genotyping arrays. The genotypes of all these samples matched those from the WGS data (average concordance rate 99.9%). 5) We also examined sample heterozygosity rate to determine any potential sample contamination. We identified three samples with increased heterozygosity rate; we assessed the ratio of allelic depth between the reference allele and alternate allele to confirm the existence of sample contamination for these three samples and then re-isolated the DNA from these samples and re-sequenced. All three samples passed QC after re-sequencing. 6) We did not observe any outliers based on duplication rate (mean 11.3%) or properly mapped rate (mean 91.5%). Additional QC for the eQTL analysis based on genotyping call rate, minor allele frequency, and HWE were described below.
CNV calling
CNVs were called using two algorithmic approaches with the goal of finding variants across a wide spectrum of sizes and making use of both read-pair and read depth information. We used the population level read-depth and split-read caller Genome STRiP (svtoolkit 2.00.1611) (Handsaker et al., 2015) to discover and genotype biallelic and multiallelic CNVs using whole genome sequencing data from 274 subjects. We merged adjacent CNVs reported by Genome STRiP using the following approach. (1) We first calculate a genotype correlation matrix for all CNVs on a given chromosome. (2) Then we create a graph where nodes are CNVs and an edge exists between two CNVs if their copy number estimates are correlated > 0.9. (3) For each connected component with more than one CNV, we sort the CNVs by position and look at each pair of adjacent CNVs and determine whether the two should be merged. We merge if the two CNVs are adjacent amongst all the calls (not just the connected component) and if the average difference in their copy number estimates is less than 0.5.
We supplemented the Genome STRiP CNV call set using the split and discordant read-pair caller LUMPY (Layer et al., 2014) as implemented in the SpeedSeq software (version 0.1.0) (Chiang et al., 2015). Speedseq SV calling was done individually on each of the 274 samples, excluding areas identified by the LUMPY developers with very high read-depth in family CEPH 1463 (Kronenberg et al., 2015). SpeedSeq CNV calls with more than 200 split or discordant reads in a given sample or that overlapped centromeres, telomeres, or low complexity regions were removed. SpeedSeq calls were then merged using svtools lsort and lmerge (Larson et al., 2016), before running the SVtyper Bayesian genotyping algorithm on these positions in each sample. Following genotyping, sites that were predicted as reference in all samples were removed as well as sites supported by less than 10 reads.
Calls from both Genome STRiP and Speedseq were removed if they overlapped the MHC region (chr6:29,600,000-33,100,000). To check the quality of our CNV calls, we investigated the concordance of calls between twins and found that Genome STRiP and SpeedSeq calls were 97% and 78% concordant between twins respectively. We also investigated the plausibility of CNV genotypes segregating in trios. Segregation plausibility was calculated using an algorithm to determine whether the observed copy number of a given CNV in a child was plausible given the observed copy numbers for the CNV in the parents. Genotypes of CNV calls segregating in 30 iPSCORE trios were found to be 99% plausible from sites discovered by Genome STRiP and 94% plausible from sites discovered by SpeedSeq.
eQTL analysis
We first selected one iPSC RNA-seq sample per subject for which WGS variant calls were also available. We constructed an empirical kinship matrix for all subjects with WGS variant calls by intersecting biallelic SNVs with 1000 Genomes phase 3 variants and LD pruning the resulting variants using plink 1.90b3x (–biallelic-only –indep-pairwise 50 5 0.2) for unrelated EUR 1000 Genomes subjects (1000 Genomes Project Consortium, 2015; Chang et al., 2015). We used the remaining LD-pruned variants to construct the kinship matrix using EPACTS 3.2.6 (epacts make-kin –min-maf 0.01 –min-callrate 0.95) keeping variants whose frequency was above 1% in our cohort and that were called in at least 95% of our samples.
We normalized RSEM gene TPM values using calcNormFactors from edgeR to account for heterogeneous, highly expressed genes that can affect the expression of genes throughout a sample (Conesa et al., 2016; Robinson and Oshlack, 2010). We then filtered the normalized TPM values by removing any genes whose expression was not greater than 2 TPM in 10 or more samples. We then transformed the expression values for each of the genes passing these filters to match a standard normal distribution and ran PEER for 15 factors (Stegle et al., 2010). After PEER, we kept only 17,805 autosomal genes and quantile normalized the PEER residuals to a standard normal to minimize the effect of outliers on the eQTL analysis (GTEx Consortium, 2015).
We filtered WGS variant calls by removing variants whose call rate was less than 95% or with Hardy-Weinberg p < 0.000001 for 104 unrelated European samples from our cohort.
We filtered GenomeSTRiP CNV calls to keep those that were observed in three diploid copy number states for at least 95% of our 215 eQTL samples. If a CNV was observed in three diploid copy number states for 95% of samples but also had other copy number states, we set those genotypes to missing. All CNVs were encoded as 0/0, 0/1, and 1/1 for increasing diploid copy number for the purposes of association. We filtered LUMPY CNV calls to keep calls with minor allele frequency greater than 1% in the 215 eQTL samples.
We tested autosomal genes for eQTLs using EMMAX (assoc –maxMAF 1 –maxMAC 1000000000 –minRSQ 0 –minCallRate 0.5 –minMAC 3) using the standard normal transformed PEER residuals and the empirical kinship matrix described above (Kang et al., 2010). We provided the sex of each subject as a covariate for EMMAX. For each gene, we tested variants within 1Mb of any TSS for that gene from the Gencode v19 gene annotation and whose call rate was greater than 95%. We identified genes with significant eQTLs (eGenes) using the permutation approach from (GTEx Consortium, 2015). For each gene, a single permutation consists of (1) permuting the expression values of the gene relative to the sample labels (e.g. we randomly assigned the expression values to different samples), (2) running EMMAX to obtain association p-values for each variant, and (3) recording the minimum p-value observed. We performed 1,000–10,000 permutations, stopping when we obtained 15 minimum p-values less than the minimum p-value observed for the real data or when we reached 10,000 permutations. We calculated an empirical p-value for each gene as the fraction of permutations with minimum p-values less than the observed minimum p-value. We corrected these empirical p-values using the Storey method (Storey and Tibshirani, 2003).
We identified additional independent eQTLs for the 5,746 eGenes by providing the lead variant as a covariate for EMMAX and performing the same permutation procedure. We corrected these permutation p-values using the Storey method and found 709 of the 5,746 eGenes had a second independent eQTL and 175 had a third eQTL.
Comparison to GTEx eQTLs
We compared our eQTLs to those reported in GTEx v6 (phs000424.v6.p1). When plotting the number of and percent unique eGenes versus the number of samples for Figure 1F–G, we omitted the GTEx testis results because they were highly different than all other GTEx tissues.
GO comparison
Genes in the “stem cell population maintenance” (GO:0019827) category were downloaded on March 17, 2016 from the AmiGO database (Ashburner et al., 2000; Carbon et al., 2009; Gene Ontology Consortium, 2015) and intersected with the 5,619 eGenes.
Functional Annotation
Roadmap Epigenomics DNase hypersensitivity site (DHS) enrichments
We downloaded DHS data for 53 Roadmap Epigenomics cell types from http://egg2.wustl.edu/roadmap/data/byFileType/peaks/consolidated/narrowPeak/. We then defined one lead variant per eGene by randomly breaking ties and keeping only SNVs and indels, resulting in 5,420 lead SNVs/indels. We removed any of the 5,420 variants that intersected a Gencode v19 exon, leaving 4,616 noncoding SNVs/indels remaining. We calculated how many SNV/indel bases did and did not overlap a DHS for each DHS experiment. We then created 5kb windows centered on each SNV/indel and calculated the number of base pairs that did and did not overlap a DHS in the window (excluding the lead variant). We used these counts to perform a Fisher exact test (fisher_exact, scipy) to determine an odds ratio and enrichment p-value for each DHS experiment as in (GTEx Consortium, 2015).
ENCODE DHS enrichments
We searched for all ENCODE DHS experiments with narrowPeak files for the hg19 assembly using the ENCODE web API (encodeproject.org) and pyencodetools (https://github.com/cdeboever3/pyencodetools). We used the most recent narrowPeak file for each experiment or chose randomly when the date was malformed. We used the same set of noncoding lead SNVs/indels described above and calculated odds ratios and enrichment p-values as described above.
ENCODE transcription factor (TF) enrichments
We identified ENCODE TF ChIP-seq experiments for the H1 hESC cell line using pyencodetools as described above. We used the same set of noncoding lead SNVs/indels described above and calculated odds ratios and enrichment p-values as described above.
Identification of putative eQTNs
To identify putative expression quantitative trait nucleotides (peQTNS) for the 5,619 eGenes, we considered all significant associations with SNVs and indels but filtered out CNV associations because their mechanism of action is likely different than disrupting a TF binding site. We also removed any eGenes that overlapped a significant CNV or had an eQTL variant that was predicted to cause nonsense mediated decay according to SnpEff (Cingolani et al., 2012). We then overlapped the remaining 186,656 variants with ENCODE TF ChIP-seq peaks (Table S4). For each variant that overlapped a peak, we calculated motif scores for motifs associated with the particular TF that the variant overlapped (http://compbio.mit.edu/encode-motifs/motifs.txt) (Kheradpour and Kellis, 2014). We calculated the motif scores using MOODS (Korhonen et al., 2009) for both the reference and alternate alleles. If the MOODS scores for the reference and alternate alleles differed by more than 2.5, we said that the variant disrupted TF binding (Table S4). When comparing to the data from (Maurano et al., 2015), we considered q < 0.05 as evidence for significant TF allelic bias.
ChIP sequencing analysis
We evaluated the quality of the 2×100 bp ChIP-seq reads using FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The reads were aligned to the hg19 reference using BWA-MEM with default parameters (Li and Durbin, 2009). Bam files were coordinate sorted using Sambamba (0.5.9) (Tarasov et al., 2015) and duplicate reads were marked using biobambam2 (2.0.21) bammarkduplicates (Tischler and Leonard, 2014). We filtered the alignments by removing reads that mapped to the ENCODE blacklist regions, had mapping quality score <30, duplicate reads, and read pairs that were not properly paired. We used MACS2 to call peaks (-g hs –keep-dup all –SPMR -s 100 –cutoff-analysis –call-summits). We merged peaks using pybedtools and counted the number of reads overlapping peaks using featureCounts (Dale et al., 2011; Liao et al., 2014; Quinlan and Hall, 2010). The counts were normalized for library size using estimateSizeFactors from DESeq2 and transformed into z-scores (Love et al., 2014).
To identify allelic binding of CTCF at peQTNs, we used ASEReadCounter (-overlap COUNT_FRAGMENTS_REQUIRE_SAME_BASE, -U ALLOW_N_CIGAR_READS) to count the number of reference and alternate alleles sequenced at each heterozygous peQTN predicted to disrupt CTCF in the five samples with ChIP-seq data. We calculated binomial p-values for each site with eight or more reads. To determine whether the direction of CTCF bias matched the predicted direction of bias based on our motif disruption predictions, we stratified the number of reads overlapping peaks that contained a CTCF peQTN by genotype: homozygous for low predicted CTCF binding, heterozygous, and homozygous for high predicted binding. We then calculated the Pearson correlation for the counts and genotype. For this analysis, we restricted to counts from peaks where the overlapped peQTN had at least one sample with significant allelic CTCF binding (p < 0.05).
Ciona embryo counting
For each experiment, once embryos had been mounted on slides, slide labels were covered with thick tape and randomly numbered by a laboratory member not involved in this project and randomized. Fifty embryos were counted for each biological replicate, unless otherwise noted. We scored embryos for expression of GFP in different embryonic tissues. Replicates were combined and p-values for differential expression between the reference and alternate alleles were calculated using a Fisher exact test.
GWAS enrichments
We downloaded the GRASP v2 database (Leslie et al., 2014). For each phenotype, we identified independent GWAS associations with p-values less than 10−5. We identified independent SNPs by creating graphs whose nodes were significant variants that shared an edge if the two variants were in LD > 0.8 (1000 Genomes phase 3 EUR). For each graph, we kept the variant with the smallest p-value per connected component and discarded the rest to create a set of independent variants. We then filtered the GRASP GWAS phenotypes to remove any phenotypes with less than 200 independent variants leaving 33 GRASP GWAS phenotypes.
To test for enrichment of GWAS associations from these 33 phenotypes among lead eQTL variants, we created 50 random sets of null SNPs matched on minor allele frequency, number of SNPs in LD > 0.8, and distance to the nearest protein coding gene; these statistics were obtained from SNPsnap (EUR population) (Pers et al., 2015). We then LD pruned eQTL lead SNVs and counted the number of independent GWAS SNPs that were in LD (LD > 0.8, 1000 Genomes phase 3 EUR) with an independent eQTL lead variant for both the real and null data. We summed the results for the 50 null sets and calculated enrichments using a Fisher exact test (fisher_exact, scipy).
CNV eQTL Analysis
CNVs eQTLs
We included GenomeSTRiP and LUMPY CNVs when mapping eQTLs as described above. While GenomeSTRiP calls multiallelic CNVs, we only used CNVs with at most three biallelic copy number states for 95% of the 215 subjects when identifying eQTLs. Mixed CNVs are defined by GenomeSTRiP as CNVs with diploid copy numbers consistent with both deletions and duplications relative to the reference. We encoded the three copy number states as 0/0, 0/1, 1/1 in order of increasing copy number for use with EMMAX. For LUMPY, we used the genotypes from SVtyper.
CNVs overlapping genes
We took a conservative approach for identifying which eGenes overlapped CNVs. We observed that in some instances, GenomeSTRiP called one CNV as two different CNVs. This was apparent because the two CNVs were in perfect LD and next to each other on the genome. We therefore merged nearby CNVs with highly correlated copy number estimates. For a given eGene, we also merged all CNVs associated with that eGene for the purpose of determining whether the eGene overlapped a significant CNV. Thus if there were two CNVs on either side of a gene and they were both associated with the expression of the gene, we would merge these two CNVs and consider that eGene to overlap a significant CNV.
CNV functional annotation
We overlapped the eQTL mCNVs with functional annotations from Roadmap Epigenomics (http://egg2.wustl.edu/roadmap/data/byFileType/peaks/consolidated/narrowPeak/) for stem cell lines ES-I3, ES-UCSF4, ES-WA7, HUES48, HUES6, HUES64, iPS, iPS-15b, iPS-18, iPS-20b. Some annotations were not available for some lines. We calculated enrichments using scipy’s fisher_exact.
Multiallelic CNV (mCNV) eQTLs
We identified mCNVs using the GenomeSTRiP CNV calls by first removing any CNVs that did not have more than three diploid copy number states among the 131 unrelated subjects or that were in the MHC region (chr6:29,600,000-33,100,000). We further filtered the mCNVs to only include mCNVs for which at least 6 subjects had diploid copy number states that differed from the three most prevalent diploid copy number states to avoid including CNVs that may have been classified as mCNVs due to erroneous copy number estimates for a small number of samples. We identified eQTLs by regressing PEER residual expression values for genes within 1Mb of an mCNV against the diploid copy number estimates for the 131 unrelateds for that mCNV. We included sex as a covariate. In total, there were 152 distinct mCNVs that we tested for eQTLs with 1,493 genes (2,952 total tests). We corrected these 2,952 test for multiple testing using the Benjamini Hochberg procedure. We determined whether an mCNV overlapped an eGene after merging the mCNVs as described above for CNVs.
Rare Variant Analysis
Rare variant identification
We first intersected GATK SNVs with promoters from Gencode v19. Promoters were defined as 2kb upstream and 200 bp downstream of a TSS for all Gencode genes. We only used promoters from 18,556 genes (including sex chromosome genes) with TPM > 2 in at least 10 of the 215 samples. We obtained DHSs for the H1, H9, iPS DF 6.9, and iPS DF 19.11 cell lines from Roadmap Epigenomics and merged them into one bed file. We the intersected the promoter variants with these merged DHSs. We next annotated each SNV with its minor allele frequency (MAF) from the 1000 Genomes phase 3. We kept variants whose MAF was less than 0.5% in all 1000 Genomes population groups and that only had one observed minor allele among the 131 unrelated individuals. We identified 65,552 rare promoter DHS SNVs (rpdSNVs) in total.
Effect of rare promoter DHS SNVs on gene expression
To determine the effect of rpdSNVs on gene expression, we focused on the expression the 17,820 genes in the 131 unrelated subjects to avoid confounding due to relatedness. We used the PEER residual gene expression estimates transformed into z-scores so that we could compare across genes. We stratified each of the 17,820×131=2,334,420 expression estimates into two groups based on whether a given gene had an rpdSNV in a given sample. In total, there were 69,013 estimates from genes/samples with an rpdSNV and 2,265,407 from genes/samples without an rpdSNV. We compared the distribution of these 69,013 and 2,265,407 expression values using a Mann Whitney U test to test whether the distributions differed. We also calculated whether a given gene/sample was more likely to have ASE if it contained an rpdSNV using a Fisher exact test.
We calculated CADD scores (Kircher et al., 2014) for all variants and used the CADD Phred scores to filter the 69,013 estimates from genes/samples with rpdSNVs to only include rpdSNVs with CADD Phred greater than 20. We also filtered based on phyloP score (Pollard et al., 2010) greater than 3.
Effect of rare genic CNVs on gene expression
We identified rare CNVs that overlapped genes where a gene was defined as the entire region from its 5′-most TSS to its 3′-most UTR. A CNV was defined as rare if it was observed in only one of the 131 unrelateds. We also characterized whether the CNV overlapped any exonic part of the gene. We stratified the 2,430,836 estimates as described above based on the presence of a genic duplication or a genic deletion. We similarly compared the distributions of expression values using a Mann Whitney U and used a Fisher exact test to test for ASE enrichment.
X Reactivation
We used 144 separate RNA-seq experiments from 116 iPSC lines derived from female subjects (some lines had biological replicates). We restricted the analysis to lines with no evidence of reprogramming-associated CNVs on the X chromosome (Panopoulos et al., In press). We used the ASE results from MBASED described above. The major haplotype frequency estimates were also produced by MBASED.
DATA AND SOFTWARE AVAILABILITY
HumanCoreExome genotyping array, RNA sequencing, ChIP sequencing, and whole genome sequencing data are available through dbGaP (phs000924 and phs001325). The whole genome sequencing genotype calls will be available at the time of publication. Due to the large file sizes of the raw whole genome sequencing reads we have not yet been able to make them publicly available, but these data will be accessible on an NHLBI cloud server via dbGaP in the near future. At that time, we will issue a correction for this manuscript with the information for retrieving the raw whole genome sequencing reads. Code for this project is available on Github at https://github.com/frazer-lab/cardips-ipsc-eqtl.
Supplementary Material
Acknowledgments
We would like to thank Terry Solomon for useful input on this project. We would like to thank James Kadonaga for gifting us the super core promoter. This work was supported in part by a CIRM grant GC1R-06673 (to KAF) and NIH grants HG008118-01 (to KAF), HL107442-05 (to KAF), and DK105541 (to KAF). We would like to thank the UC San Diego Institute for Genomic Medicine (IGM) core for sequencing services with support from NIH grant P30CA023100. CD is supported in part by the University of California, San Diego, Genetics Training Program through an institutional training grant from the National Institute of General Medical Sciences (T32GM008666) and the California Institute for Regenerative Medicine (CIRM) Interdisciplinary Stem Cell Training Program at UCSD II (TG2-01154). DJ is supported by National Library of Medicine Training Grant 4T15LM011271-05. PB is supported by the Swiss National Science Foundation (SNSF) P2LAP3-155105. Equipment in the Neuroscience Microscopy core was used for the Ciona experiments; this core is supported by the UCSD School of Medicine Microscopy Core Grant P30 NS047101. “Man” and “Woman” logos for graphical abstract were created by delicti from thenounproject.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
CD designed and performed computational analyses. CD and KAF designed experiments. HL performed WGS alignment and GATK variant calling. DJ performed GenomeSTRiP and LUMPY CNV calling. ADC, AA, PB performed iPSC cell culture and collected RNA for sequencing. WB and ES generated whole genome sequencing data. CD, HM, ENS, MA, NN, and JR processed data and organized data in a database. HH, KJ, and BR designed and performed ChIP-seq experiments. KMO and EKF performed Ciona experiments and analyzed the data. CD, EKF, and KAF wrote the manuscript. All authors edited the manuscript.
References
- 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abitua PB, Gainous TB, Kaczmarczyk AN, Winchell CJ, Hudson C, Kamata K, Nakagawa M, Tsuda M, Kusakabe TG, Levine M. The pre-vertebrate origins of neurogenic placodes. Nature. 2015;524:462–465. doi: 10.1038/nature14657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet. 2015;16:197–212. doi: 10.1038/nrg3891. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol. 2016;17:170–182. doi: 10.1038/nrm.2015.27. [DOI] [PubMed] [Google Scholar]
- Banovich NE, Li YI, Raj A, Ward MC, Greenside P, Calderon D, Tung PY, Burnett JE, Myrthil M, Thomas SM, et al. Impact of regulatory variation across human iPSCs and differentiated cells. bioRxiv. 2016:1–17. doi: 10.1101/gr.224436.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S, Ami GOH, Web Presence Working, G AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;25:288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Ayyanathan K. Snail/Gfi-1 (SNAG) family zinc finger proteins in transcription regulation, chromatin dynamics, cell signaling, development, and disease. Cytokine Growth Factor Rev. 2013;24:123–131. doi: 10.1016/j.cytogfr.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, Marth GT, Quinlan AR, Hall IM. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods. 2015;12:966–968. doi: 10.1038/nmeth.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Scott AJ, Davis JR, Tsang EK, Li X, Kim Y, Damani FN, Ganel L, GTEx Consortium. Montgomery SB, et al. The impact of structural variation on human gene expression. bioRxiv. 2016:1–26. doi: 10.1038/ng.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Lee S, Mallard W, Clement K, Tagliazucchi GM, Lim H, Choi IY, Ferrari F, Tsankov AM, Pop R, et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat Biotechnol. 2015;33:1173–1181. doi: 10.1038/nbt.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiaen L, Wagner E, Shi W, Levine M. Electroporation of transgenic DNAs in the sea squirt Ciona. Cold Spring Harb Protoc. 2009;2009 doi: 10.1101/pdb.prot5345. pdb prot5345. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szczesniak MW, Gaffney DJ, Elo LL, Zhang X, et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17:13. doi: 10.1186/s13059-016-0881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale RK, Pedersen BS, Quinlan AR. Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics. 2011;27:3423–3424. doi: 10.1093/bioinformatics/btr539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delsuc F, Brinkmann H, Chourrout D, Philippe H. Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature. 2006;439:965–968. doi: 10.1038/nature04336. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erives A, Corbo JC, Levine M. Lineage-specific regulation of the Ciona snail gene in the embryonic mesoderm and neuroectoderm. Dev Biol. 1998;194:213–225. doi: 10.1006/dbio.1997.8810. [DOI] [PubMed] [Google Scholar]
- Farley EK, Olson KM, Zhang W, Brandt AJ, Rokhsar DS, Levine MS. Suboptimization of developmental enhancers. Science. 2015;350:325–328. doi: 10.1126/science.aac6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney DJ, Veyrieras JB, Degner JF, Pique-Regi R, Pai AA, Crawford GE, Stephens M, Gilad Y, Pritchard JK. Dissecting the regulatory architecture of gene expression QTLs. Genome Biol. 2012;13:R7. doi: 10.1186/gb-2012-13-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Nicolae DL, Cox NJ. A study of CNVs as trait-associated polymorphisms and as expression quantitative trait loci. PLoS Genet. 2011;7:e1001292. doi: 10.1371/journal.pgen.1001292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Stranger BE. The impact of human copy number variation on gene expression. Brief Funct Genomics. 2015;14:352–357. doi: 10.1093/bfgp/elv017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, McCarroll SA. Large multiallelic copy number variations in humans. Nat Genet. 2015;47:296–303. doi: 10.1038/ng.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Dadon DB, Powell BE, Fan ZP, Borges-Rivera D, Shachar S, Weintraub AS, Hnisz D, Pegoraro G, Lee TI, et al. 3D Chromosome Regulatory Landscape of Human Pluripotent Cells. Cell Stem Cell. 2016;18:262–275. doi: 10.1016/j.stem.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juven-Gershon T, Cheng S, Kadonaga JT. Rational design of a super core promoter that enhances gene expression. Nat Methods. 2006;3:917–922. doi: 10.1038/nmeth937. [DOI] [PubMed] [Google Scholar]
- Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, Sabatti C, Eskin E. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradpour P, Kellis M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 2014;42:2976–2987. doi: 10.1093/nar/gkt1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpinen H, Goncalves A, Leha A, Afzal V, Ashford S, Bala S, Bensaddek D, Casale FP, Culley O, Danacek P, et al. Common genetic variation drives molecular heterogeneity in human iPSCs. bioRxiv. 2016:1–23. doi: 10.1038/nature22403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen J, Martinmaki P, Pizzi C, Rastas P, Ukkonen E. MOODS: fast search for position weight matrix matches in DNA sequences. Bioinformatics. 2009;25:3181–3182. doi: 10.1093/bioinformatics/btp554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg ZN, Osborne EJ, Cone KR, Kennedy BJ, Domyan ET, Shapiro MD, Elde NC, Yandell M. Wham: Identifying Structural Variants of Biological Consequence. Plos Comput Biol. 2015;11:e1004572. doi: 10.1371/journal.pcbi.1004572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedlander MR, t Hoen PA, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson D, Abelh J, Chiang C, Badve Abhijit, Morton D, Eldred J. svtools: svtools v0.2.0a1 2016 [Google Scholar]
- Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84. doi: 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie R, O’Donnell CJ, Johnson AD. GRASP: analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics. 2014;30:i185–194. doi: 10.1093/bioinformatics/btu273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessing D, Anguera MC, Lee JT. X chromosome inactivation and epigenetic responses to cellular reprogramming. Annu Rev Genomics Hum Genet. 2013;14:85–110. doi: 10.1146/annurev-genom-091212-153530. [DOI] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Battle A, Karczewski KJ, Zappala Z, Knowles DA, Smith KS, Kukurba KR, Wu E, Simon N, Montgomery SB. Transcriptome sequencing of a large human family identifies the impact of rare noncoding variants. American journal of human genetics. 2014;95:245–256. doi: 10.1016/j.ajhg.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Kim Y, Tsang EK, Davis JR, Damani FN, Chiang C, Zappala Z, Strober BJ, Scott AJ, Ganna A, et al. The impact of rare variation on gene expression across tissues. bioRxiv. 2016 doi: 10.1038/nature24267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liu X, Sun H, Qi J, Wang L, He S, Liu J, Feng C, Chen C, Li W, Guo Y, et al. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT-MET mechanism for optimal reprogramming. Nat Cell Biol. 2013;15:829–838. doi: 10.1038/ncb2765. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.) Nature. 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- Maurano MT, Haugen E, Sandstrom R, Vierstra J, Shafer A, Kaul R, Stamatoyannopoulos JA. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat Genet. 2015;47:1393–1401. doi: 10.1038/ng.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayba O, Gilbert HN, Liu J, Haverty PM, Jhunjhunwala S, Jiang Z, Watanabe C, Zhang Z. MBASED: allele-specific expression detection in cancer tissues and cell lines. Genome Biol. 2014;15:405. doi: 10.1186/s13059-014-0405-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan R, Watt FM. What is the point of large-scale collections of human induced pluripotent stem cells? Nat Biotechnol. 2013;31:875–877. doi: 10.1038/nbt.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, et al. Human genomics. The human transcriptome across tissues and individuals. Science. 2015;348:660–665. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai AA, Pritchard JK, Gilad Y. The genetic and mechanistic basis for variation in gene regulation. PLoS Genet. 2015;11:e1004857. doi: 10.1371/journal.pgen.1004857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pala M, Zappala Z, Marongiu M, Li X, Davis JR, Cusano R, Crobu F, Kukurba KR, Reiner F, Berutti R, et al. Population and individual effects of non-coding variants inform genetic risk factors. bioRxiv 2016 [Google Scholar]
- Panopoulos AD, D’Antonio M, Benaglio P, Williams R, Hashem SI, Schuldt BM, DeBoever C, Arias AD, Garcia M, Nelson B, et al. iPSCORE: A systematically derived resource of iPSC lines from 222 individuals for use in examining how genetic variation affects molecular and physiological traits across a variety of cell types. Stem Cell Reports. doi: 10.1016/j.stemcr.2017.03.012. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasque V, Plath K. X chromosome reactivation in reprogramming and in development. Curr Opin Cell Biol. 2015;37:75–83. doi: 10.1016/j.ceb.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pers TH, Timshel P, Hirschhorn JN. SNPsnap: a Web-based tool for identification and annotation of matched SNPs. Bioinformatics. 2015;31:418–420. doi: 10.1093/bioinformatics/btu655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouhani F, Kumasaka N, de Brito MC, Bradley A, Vallier L, Gaffney D. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 2014;10:e1004432. doi: 10.1371/journal.pgen.1004432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegle O, Parts L, Durbin R, Winn J. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. Plos Comput Biol. 2010;6:e1000770. doi: 10.1371/journal.pcbi.1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolfi A, Ryan K, Meinertzhagen IA, Christiaen L. Migratory neuronal progenitors arise from the neural plate borders in tunicates. Nature. 2015;527:371–374. doi: 10.1038/nature15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streeter I, Harrison PW, Faulconbridge A, The HipSci, C. Flicek P, Parkinson H, Clarke L. The human-induced pluripotent stem cell initiative-data resources for cellular genetics. Nucleic Acids Res. 2017;45:D691–D697. doi: 10.1093/nar/gkw928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–2034. doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchieu J, Kuoy E, Chin MH, Trinh H, Patterson M, Sherman SP, Aimiuwu O, Lindgren A, Hakimian S, Zack JA, et al. Female human iPSCs retain an inactive X chromosome. Cell Stem Cell. 2010;7:329–342. doi: 10.1016/j.stem.2010.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SM, Kagan C, Pavlovic BJ, Burnett J, Patterson K, Pritchard JK, Gilad Y. Reprogramming LCLs to iPSCs Results in Recovery of Donor-Specific Gene Expression Signature. PLoS Genet. 2015;11:e1005216. doi: 10.1371/journal.pgen.1005216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler G, Leonard S. biobambam: tools for read pair collation based algorithms on BAM files. Source Code for Biology and Medicine. 2014;9:13–13. [Google Scholar]
- Tsankov AM, Akopian V, Pop R, Chetty S, Gifford CA, Daheron L, Tsankova NM, Meissner A. A qPCR ScoreCard quantifies the differentiation potential of human pluripotent stem cells. Nat Biotechnol. 2015;33:1182–1192. doi: 10.1038/nbt.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UK 10K Consortium. The UK10K project identifies rare variants in health and disease. Nature. 2015;526:82–90. doi: 10.1038/nature14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Geijn B, McVicker G, Gilad Y, Pritchard JK. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat Methods. 2015;12:1061–1063. doi: 10.1038/nmeth.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;11:11 10 11–11 10 33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyrieras JB, Kudaravalli S, Kim SY, Dermitzakis ET, Gilad Y, Stephens M, Pritchard JK. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Luca F, Pique-Regi R. Cross-population joint analysis of eQTLs: fine mapping and functional annotation. PLoS Genet. 2015;11:e1005176. doi: 10.1371/journal.pgen.1005176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Enge M, Whitington T, Dave K, Liu J, Sur I, Schmierer B, Jolma A, Kivioja T, Taipale M, et al. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell. 2013;154:801–813. doi: 10.1016/j.cell.2013.07.034. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Wang G, Yang E, Ji G, Brinkmeyer-Langford CL, Cai JJ. Aberrant gene expression in humans. PLoS Genet. 2015;11:e1004942. doi: 10.1371/journal.pgen.1004942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Akinsanmi I, Arafat D, Cradick TJ, Lee CM, Banskota S, Marigorta UM, Bao G, Gibson G. A Burden of Rare Variants Associated with Extremes of Gene Expression in Human Peripheral Blood. American journal of human genetics. 2016;98:299–309. doi: 10.1016/j.ajhg.2015.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.