

Abstract
The icosahedron and the dodecahedron are the largest of the Platonic solids, and icosahedral protein structures are widely utilized in biological systems for packaging and transport1,2. There has been considerable interest in repurposing such structures3–5, for example, virus-like particles for the targeted delivery and vaccine design. The ability to design proteins that self assemble into precisely specified, highly ordered icosahedral structures would open the door to a new generation of protein 'containers' that could exhibit properties custom-made for various applications. In this manuscript, we describe the computational design of an icosahedral nano-cage that self-assembles from trimeric building blocks. Electron microscopy images of the designed protein expressed in E. coli reveals a homogenous population of icosahedral particles nearly identical to the design model. The particles are stable in 6.7 M guanidine hydrochloride at up to 80 °C, and undergo extremely abrupt, but reversible, disassembly between 2 M and 2.25 M guanidinium thiocyanate. The icosahedron is robust to genetic fusions: one or two copies of superfolder GFP can be fused to each of the 60 subunits to create highly fluorescent standard candles for light microscopy, and a designed protein pentamer can be placed in the center of each of the twenty pentameric faces to potentially gate macromolecule access to the nanocage interior. Such robust designed nanocages should have considerable utility for targeted drug delivery6, vaccine design7, and synthetic biology8.
Programming protein subunits to self-assemble into well-defined complexes is a promising route to the custom design of macromolecular machines. Protein assemblies have been engineered using metals9,10, disulfide bonds11–14, genetic fusions12,15–17, and ideal helix-helix interactions11,13,16, but these approaches have generally yielded polydisperse or unanticipated products. Recently, symmetric modeling coupled with computational protein-protein interface design has accurately generated protein assemblies with tetrahedral and octahedral symmetry18,19, but these relatively small (<16 nm diameter) nanocages have limited use for packaging or delivery applications as they have little internal volume.
Icosahedral point group symmetry contains two-, three-, and five-fold axes of rotation (Figure 1a). To generate novel icosahedral protein assemblies, trimeric protein scaffolds of known structure were arranged with icosahedral symmetry (the 3 fold axes of the trimers aligned with the 3 fold axes of an icosahedron) and the two remaining degrees of freedom — the distance (r) from the icosahedron center to the center of mass of each trimer, and the angle (ω) of rotation of each trimer about its axis — were optimized for close packing without steric clashes (Figure 1b–c). The amino acid sequences at the newly formed interfaces between the trimer building blocks were then optimized using RosettaDesign20,21, and 17 designs were selected for experimental characterization based on properties of the designed interface, including shape complementarity22, predicted binding energy, and the number of buried unsatisfied hydrogen bond donors and acceptors (see Methods).
Figure 1. Design methodology and biochemical characterization.

a–b) Icosahedral 3-fold axis in red and aligned trimeric building block in green. c) Optimization of r and ω yields closely opposed interfaces between subunits. d) Sequence design yields low energy interfaces; in the I3-01 case, composed of 5 designed residues (thick) and 2 native residues (thin). e) I3-01 appears larger by SEC than the similarly sized I3-01(L33R) and wild type trimer (1wa3). f) DLS measurement of hydrodynamic radius of 1wa3 (3.5 nm) and I3-01 (14 nm). I3-01 remains assembled in 6.7 M GuHCl and in 2 M GITC. g) Extremely sharp disassociation to trimeric building blocks at 2.25 M GITC. Data points represent independent measurements. h) I3-01 icosahedron disassembles into the trimeric building blocks at 3 M GITC, and reassembles following dilution to 1 M.
Genes encoding the designs were assembled from oligonucleotides and cloned into pET29b+ for expression in E. coli. Most of the designs were found in the insoluble fraction upon cell lysis; of the three soluble designs, two (both based on a KDPG aldolase23,24) showed significant shifts in migration relative to the wild-type scaffold when analyzed by native (non-denaturing) PAGE, suggesting higher order assembly. We selected the one with fewer mutations, I3-01, with four non-polar substitutions generating the new designed interface between trimers (E33L, L61M, N187V, and R190A) for further analysis (Figure 1d; amino acid sequences provided in Supplementary Text).
I3-01 was purified using immobilized metal affinity and size exclusion chromatography (SEC), yielding a single peak with an apparent molecular weight much larger than the wild-type trimeric protein and consistent with the expected elution volume for the 60-subunit assembly (Figure 1e). A mutant bearing a leucine to arginine substitution predicted to disrupt the designed interface eliminated the high molecular weight species and returned the elution volume to that of the wild-type scaffold (Figure 1e). Dynamic light scattering (DLS) measurements of I3-01 showed a monodisperse population of particles with a hydrodynamic radius of 14 nm, consistent with the design model (Figure 1f). No disassembly to the trimeric building block was observed at 80 °C or, remarkably, in 6.7 M GuHCl (Extended Data Figure 1). This hyperstability is a property of both the trimeric building block and the designed interface: both are completely resistant to GuHCl denaturation. An exceptionally sharp disassociation into the constituent trimers was observed between 2 and 2.25 M GITC: at 2 M the dominant species is the icosahedron, while at 2.25 M only the trimeric building block is observed (Figure 1g, Extended Data Figure 2). Importantly for cargo packaging applications, the disassociation is fully reversible: the hydrodynamic radius of particles formed by diluting disassembled protein in 3 M GITC down to 1 M GITC is identical to those originally produced in E. coli (Figure 1h).
The structure of I3-01 was investigated using cryo-electron microscopy (cryo-EM). Micrographs of individual particles were highly homogenous (Figure 2a), and in class averages from 6,461 particles, the three projections along the symmetry axes and the overall icosahedral architecture are clearly discernible (Figure 2b–c). A 3D model calculated from the cryo-EM data matches the I3-01 design model very well with a correlation coefficient of 0.92 at 20 Å and 1.5 σ (Figure 2d–e), clearly indicating that I3-01 forms the designed structure: an icosahedron with a diameter of 25 nm and an interior volume of approximately 3,000 nm3, values that are in range of those observed in small viral capsids25.
Figure 2. Cryo-Electron Microscopy.

a) Field of view cryo-EM micrograph with homogeneous icosahedral particles in various orientations. b) Back projections of I3-01 from the design model. c) CryoEM class averages closely match the design projections along all three symmetry axes. d–e) The calculated density (blue, 3.22 σ) closely matches the design model (green).
To probe the robustness of I3-01 to genetic fusion, we fused superfolder GFP (sfGFP)26 to one or both termini of the monomeric subunit and produced the resulting proteins in E. coli. SEC analysis showed that the fusion proteins had hydrodynamic radii consistent with cage formation (Extended Data Figure 3). Analysis of I3-01 with a C-terminal sfGFP fusion (I3-01(ctGFP)) by cryo-EM revealed icosahedral particles with very similar overall shape to the original design. Class averages of 13,297 particles revealed considerable internal density compared to the original I3-01 averages, consistent with the computational model of the fusion complex (Figure 3a). The I3-01 sfGFP fusions are robust to denaturation of the N- or C- terminal fused sfGFP in GuHCl; the particles remain assembled as GFP signal is lost27 (Extended Data Figure 4).
Figure 3. Tuning nanocage structure and function with genetic fusions.

a) (left) Cryo-EM micrograph of I3-01(ctGFP). (top right) Computational model; sfGFP in green. (bottom right) Class average along the 5-fold axis. b) Fluorescence microscopy fields of view, c) fluorescence intensity histograms, and d) correlation between the mean fluorescence intensity and sfGFP copy number for nanoparticles with different numbers of fused sfGFP molecules. e–f) Computational model and class averages along the five fold axis of negatively stained e) I3-01 and f) I3-01(HB); helical bundle in red. Three class averages are shown as there is some heterogeneity at the pentameric face. Weak density in the center of the pentameric faces in I3-01 may reflect randomly packaged material. There is clear density in the center of the pentameric faces in the I3-01(HB) class averages consistent with the model.
It is currently challenging to infer subunit copy number in GFP tagged assemblies from their fluorescence intensity. What is needed are “standard candles” with known fluorescent protein copy numbers that can be used to correlate fluorescence intensity to copy number. To complement the icosahedra with 60 and 120 copies of sfGFP described above, we fused sfGFP to one or both components of a previously described two-component tetrahedron (T33-2119) to generate assemblies with 12 or 24 copies of sfGFP (Extended Data Figure 3). Intensity histograms obtained for each of the sfGFP-nanocage constructs using widefield fluorescence microscopy were well fit with Gaussians (Figure 3b–c), and the mean fluorescence intensity for each cage was found to be linearly proportional (r2 = 0.9925) to sfGFP copy number (Figure 3d). The fluorescent properties of the particles were readily manipulated by substituting sfGFP with mTurqoise2 and sYFP2 (Extended Data Figure 5). In addition to serving as genetically encoded, water-soluble fluorescent standard candles, the fluorescent protein cage fusions could be useful for correlative light and electron microscopy28 as the icosahedral shape is quite distinctive.
In I3-01, the trimeric building blocks are aligned with the three-fold axes while the designed interface is along the icosahedral two-folds. To explore the possibility of symmetry-matched fusions to designed nanocages, we modeled a designed pentameric helical bundle29 into the center of the large 9 nm pore at the five-fold axis with a C-terminal loop fusion (I3-01(HB)). Negative stain electron microscopy showed monodisperse particles of the expected size and symmetry; the incorporation of the pentamer does not interfere with icosahedron assembly. Particle averages showed a structure similar to that of the original icosahedron, with additional density at the center of each five-fold axis, consistent with the design model (Figure 3e–f). The capability of incorporating symmetry-matched substructures into designed nanocages offers considerable flexibility and modularity; for example, pentamers filling otherwise open pentameric faces could gate release of cargo contained within the nanocage.
The designed I3-01 icosahedron is exceptionally stable, robust to genetic fusion, and has a considerably larger internal volume than previously designed nanocages with well-defined and prespecified structures14,17,19. Enzymatic activity is retained in the assembled icosahedron (Extended Data Figure 6), suggesting a route to custom nanoreactors. The ability to accurately design icosahedral protein structures also opens the door to new approaches to vaccine generation and targeted drug delivery.
Methods
Computational Design
Crystal structures of 300 trimers were selected from the Protein Data Bank (PDB) to use as building blocks. These structures were selected based on their resolution (<2.5 Å) and “designability”, excluding scaffolds with many loops or flexible domains on potential design surfaces. For each scaffold, 20 trimeric building blocks were arranged in icosahedral symmetry by aligning the 3-fold rotational axis of each trimer with one of the 3-fold icosahedral symmetry axes. While preserving symmetry, the building blocks were then docked together by enumeratively sampling their rotations (ω) about the 3-fold symmetry axes and translating (r) them into contact along the aligned axes. Configurations were discarded in which backbone atoms from different building blocks were less than or equal to 3.5 Å apart. Each configuration was scored based on the number of β-carbon contacts between adjacent building blocks (where a contact is defined as two β-carbons within a distance of 12 Å), with docking data recorded for up to 30 top scoring configurations for each scaffold. A total of 208 docked configurations were selected for interface design. The selected configurations were re-sampled more finely in 0.5° and 0.2Å increments, and symmetric RosettaDesign20,21 was applied to mutate relevant interface residues to generate low-energy and symmetric hydrophobic interfaces. These designs were then filtered based on a series of Rosetta score values such as shape complementarity22 (sc), interface surface area (sasa), buried unsatisfied hydrogen bonds (uhb), and binding energy (ddg). All mutations in passing designs were then analyzed by rescoring each mutated residue relative to its wild-type counterpart as a measure of relevancy. All final designs were inspected manually before experimental characterization. The full 60-subunit design model of I3-01 is provided as a supplementary file.
Cloning, screening, and Protein Purification
Codon-optimized genes encoding the wild type and designs were generated by recursive PCR from sets of synthetic oligonucleotides (Integrated DNA Technologies). Five mutations were incorporated into I3-01: E26K, E33L, K61M, D187V, and R190A. All genes were cloned into the pET29b+ plasmid with kanamycin resistance and expressed in BL21 Star (DE3) E. coli cells (Invitrogen) induced with IPTG for 4 hours at 37 °C. Cell lysis was accomplished in TBS (50 mM Tris, 500 mM NaCl) with lysozyme (0.25 mg/mL) and sonication (Fisher Scientific) at 20W for 5 minutes total on time, using 10/10 seconds on/off rounds.
For initial screening, all constructs were labeled with the CoA-488 flourophore (NEB) by the addition of AcpS30 (NEB) using an A1 peptide tag, allowing the solubility and assembly state of each design to be analyzed using SDS-PAGE and native-PAGE (BIO-RAD), following procedures previously described18. All subsequent experiments were performed on either (His)6-tagged protein or remained untagged.
After lysis and centrifugation at 20,000× G for 30 minutes, the soluble fraction of (His)6-tagged proteins were passed through 2 mL of Ni-NTA (Qiagen), washed with 30 mM imidazole, and eluted with 500 mM imidazole. Pure proteins were collected after elution from a Superose 6 10/300 GL SEC column (GE Healthcare) at 9–11 mL, depending on the fusion variant.
Non-(His)6-tagged proteins were lysed identically, and then the cleared lysates were treated with serial ammonium sulfate precipitation treatments (20%, 60% w/v). During each step, solid ammonium sulfate was added to the lysate to the desired percentage, and equilibrated at room temperature for 1 hour. Ammonium sulfate precipitated protein was then collected by centrifugation at 20,000× G for 30 minutes at 25 °C. After treatment at 60%, the pellet was then solubilized in TBS and heated at 80 °C for 10 minutes. The soluble fraction was then collected and further purified through SEC as described.
KDPG Enzyme Assay
The reaction was carried out in 25 mM HEPES, 20 mM NaCl buffer at pH 7.0 with the presence of NADH (0.1 mM), L-lactate dehydrogenase (LDH, 0.11 U/µL), and 2-keto-3-deoxy-6-phosphogluconate (KDPG, 1 mM) at 25°C, based on previously described methods23. Native 1wa3, I3-01, or I3-01(K129A) was added at 0.02 µM final concentration to each well and immediately monitored for 339 nm UV absorbance over time.
Dynamic Light Scattering
Purified protein was measured using a DynaPro NanoStar (Wyatt) dynamic light scattering (DLS) setup. 0.5 mg/mL of I3-01 and 1wa3 were measured at 25 °C, then the temperature was ramped up to 90 °C, then ramped back down to 25 °C for temperature scans at 2 °C/minute. Measurements were taken in the presence of Tris-buffered saline (TBS): 25mM Tris, 500mM NaCl; buffered guanidinium hydrochloride (GuHCl): 25mM Tris, 500mM NaCl, 1–6.7 M GuHCl; or buffered guanidine thiocyanate (GITC): 25mM Tris, 500mM NaCl, 1–4 M GITC. Different concentrations of GITC equilibrated samples were achieved by combining stocks of 0 M and 4 M equilibrated solutions in different ratios while GuHCl equilibrated samples were equilibrated individually. Each sample was allowed to equilibrate in their respective buffer for at least 24 hours before measurement. Re-annealing experiments were performed by diluting I3-01 equilibrated in 3 M GITC down to 1 M GITC final concentration (0.166 mg/mL protein). Data analysis was performed using DYNAMICS v7 (Wyatt), reporting regularization fits (with D10/D50/D90) except for temperature ramp experiments, where cumulant fits were used. The ~1 nm radius particle consistent with GITC buffer alone was disregarded for analysis, and monodispersity was assumed when peak polydispersity was below 15%31,32.
Negative-Stain Electron Microscopy
3 µl of purified I3-01 and I3-01(ctGFP) at 0.1 mg/mL were applied to glow discharged, carbon coated 200-mesh copper grids (Ted Pella, Inc.), washed with Milli-Q water and stained with 0.75% uranyl formate as described previously33. Grids were visualized for assembly validation and stability and subsequently optimized for Cryo-EM data collection. Screening was performed on a 120 kV Tecnai Spirit T12 transmission electron microscope (FEI) with a bottom-mount TVIPS F416 CMOS 4k camera.
6 µl of purified I3-01 and I3-01(HB) at 0.05 to 0.1 mg/mL were applied to glow discharged, carbon coated 400-mesh copper grids (Ted Pella, Inc.), washed with Milli-Q water and stained with 0.75% uranyl formate. Grids were visualized for assembly validation and optimized for data collection. Screening and sample optimization was performed on a 100kV Morgagni M268 transmission electron microscope (FEI) equipped with an Orius CCD camera (Gatan). Data collection was performed on a 120 kV Tecnai G2 Spirit transmission electron microscope (FEI). All final images were recorded using an Ultrascan 4000 4k × 4k CCD camera (Gatan) at 52,000× magnification at the specimen level. Coordinates for 6,576 I3-01 and 11,726 I3-01(HB) unique particles were obtained for averaging using EMAN234. Boxed particles were used to obtain 2D class averages by refinement in EMAN2. Additional image analysis was performed using ImageJ35.
Cryo Electron Microscopy
5 µl of purified untagged I3-01 and I3-01(ctGFP), diluted to ~0.1 mg/mL using TBS buffer (25 mM Tris pH 8.0, 150mM NaCl) with an additional 2 mM Dithiothreitol were applied to glow discharged 1.2/1.3 Quantifoil grids, blotted and plunged into liquid ethane using a Vitrobot (FEI). Screening and grid optimization was performed on a 200 kV TF20 transmission electron microscope (FEI) with a bottom-mount TVIPS F416 CMOS 4k camera. 4–6 second movies were recorded on a 300 kV Titan Krios (FEI) using a Gatan K2 direct detector at either 29,000× or 37,000× magnification at the specimen level at ~10 electrons/pixel/second.
Movies were motion corrected using previously described methods36. Coordinates for 6,461 (I3-01) and 13,297 (I3-01(ctGFP)) unique particles were obtained for averaging using EMAN234. Extracted frames of these particles were used to calculate class averages by refinement in IMAGIC37 using multiple rounds of MSA (multivariate statistical analysis) and MRA (multi-reference alignment). An initial density model was calculated based on the calculated averages using EMAN234 and the fitting of the model and correlation calculated using UCSF Chimera38. Low-resolution (17–30 Å) volumes from the I3-01 design model were calculated using SPIDER39 and inspected in UCSF Chimera38. Back-projection images were computed in SPIDER39 on the low-resolution volumes and visualized using WEB39. The contrast of all micrographs was enhanced in Fiji40.
Symmetrical Linker Modeling
RosettaRemodel41 was used to model I3-01(ctGFP) and to generate ideal linker lengths for I3-01(HB). For I3-01(ctGFP), I3-01 was held static while the linker was sampled via fragment insertion, placing the sfGFP molecules at the end of the linker. The overall model was sampled symmetrically with icosahedral symmetry.
For I3-01(HB), I3-01 was held static while linkers of different lengths (7–12 residues) were sampled via fragment insertion. The resulting placement of the helical bundle at the end of the linker was filtered with pentameric assembly constraints to determine linker lengths that could satisfy formation of the pentameric helical bundle. The shorter linkers that allowed unstrained helical assembly were selected for experimental testing.
Fluorescence microscopy
Different constructs used for fluorescence microscopy were generated by genetically fusing sfGFP to the termini of nanocages. For T33-21, the sfGFP was fused to either the C-terminus of the first component (12 sfGFP molecules), or the C-terminus of both components (24 sfGFP molecules). For I3-01, the sfGFP was fused to either termini of I3-01 (60 sfGFP molecules), or both (120 sfGFP molecules). For mTurquoise2 and SYFP2 versions, sfGFP was replaced with the sequence of the respective fluorescent protein bearing additional surface mutations identical to sfGFP26.
GFP nanocages were mounted on agarose pads for microscopy as previously described42. Images of the GFP nanocages were obtained using a DeltaVision system (Applied Precision) with an IX70 inverted microscope (Olympus), a U Plan Apo 100× objective (1.35 NA) and a CoolSnap HQ digital camera (Photometrics). GFP images were taken with a 0.4 second exposure, in a single focal plane, and binned 1×1. The fluorescence intensities of GFP puncta were identified and quantified using custom Matlab programs previously described43. Fluorescent intensity histograms of individual sfGFP-fused cages were fit with Gaussian distributions, shown with mean total arbitrary fluorescence unit (AFU) intensity ± std.dev.
Extended Data
Extended Data Figure 1. I3-01 Tolerance to Temperature.

Dynamic light scattering measurements as I3-01 is subjected to heating to 90°C (solid line), then cooling to 25°C (dotted line) in a) TBS, b) 6.7 M GuHCl, and c) 2 M GITC. In all 3 conditions, any indications of aggregation or increase in size due to temperature appear to be completely reversible.
Extended Data Figure 2. Reproducibility of I3-01 transition in 2 to 2.25 M GITC.

Four examples each of independent measurements at 2 M (blue) and 2.25 M (red) GITC using DLS show the reproducibility of the cage disassociation. Histograms are plotted offset by 1% intensity from each other for clarity.
Extended Data Figure 3. SEC of T33-21 and I3-01 fused with sfGFP.

Size exclusion chromatography traces for T33-21 (12-mer in red and 24-mer in blue) and I3-01 (60-mer in green and 120-mer in purple) sfGFP fusions, display increased particle sizes with increasing copies of GFP, but retain monodispersed populations. The N-terminal fusion of sfGFP (dashed line) is expected to extend mostly outwards of the icosahedron, thus greatly increasing the hydrodynamic radius while the C-terminal fusion is predicted to occupy the internal void space.
Extended Data Figure 4. Tolerance of I3-01-sfGFP fusions to GuHCl.

N-terminal (red) and C-terminal (blue) sfGFP fusions were equilibrated to 0–6.4 M GuHCl. UV absorbance at 490 nm monitors the unfolding of sfGFP (top, solid line). DLS experiments (top, dotted line) reveal as sfGFP unfolds, the hydrodynamic radius increases slightly, and then stabilizes. Bottom panels show that in 1 M GuHCl (solid line) and in 6 M GuHCl (dotted line), the icosahedral assemblies remain relatively monodisperse.
Extended Data Figure 5. I3-01 C-terminal fusions with other fluorescent proteins.

Fluorescent proteins mTurquoise2 or sYFP2 were fused to the C-terminus of I3-01. The field of view using widefield fluorescence microscopy shows distinct signals of each type when the two types are mixed together.
Extended Data Figure 6. I3-01 Retains Native Enzyme Activity.
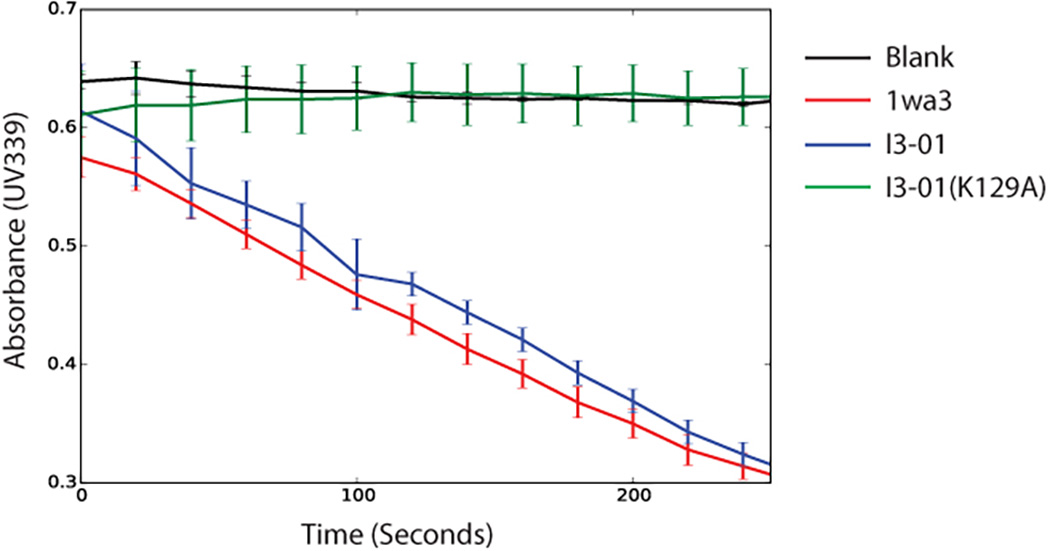
Coupled KDPG aldolase assay showing native-like enzymatic activity in I3-01. The K129A knockout shows no enzyme activity, similar to buffer alone.
Supplementary Material
Acknowledgments
This work was supported by the Howard Hughes Medical Institute (D.B. and T.G.), the JRC visitor program (S.G.), the National Science Foundation CHE-1332907 (D.B.), a UW/Hutch CCSG Pilot Award NCI 5 P30 CA015704-41 (D.B. and N.P.K.), Takeda Pharmaceutical Company (N.K.), and the Bill and Melinda Gates Foundation OPP1120319 (D.B. and N.K), and the National Institute of Health P41 GM103533 (T.N.D.). Y.H. was supported in part by a NIH Molecular Biology Training Grant (T32GM008268). U.N. was supported in part by PHS National Research Service Award (T32GM007270) from NIGMS. J.B.B. was supported in part by an NSF Graduate Research Fellowship (DGE-0718124). We would like to thank the Janelia Research Campus Cryo-EM Facility and Jason de la Cruz for their assistance with the Titan Krios.
Footnotes
Author Contributions
J.B.B., N.P.K., and W.S. developed the computational design methodology. Y.H. and J.B.B. performed the design of the icosahedra. Y.H. performed all other unlisted experiments. S.G. and D.S. performed the cryo-EM experiments. K.K.F. performed the fluorescence microscopy experiments. U.N. performed the negative stain EM experiments. C.X. provided the pentamer sequence for I3-01(HB). P.H. provided the computational methodology to model fusions to I3-01. R.R. produced I3-01(HB) proteins. S.Y. produced T33-21 sfGFP fusion proteins.
The authors have no competing interests as defined by Nature Publishing Group, or other interests that might be perceived to influence the results and/or discussion reported in this article.
References
- 1.Zandi R, Reguera D, Bruinsma RF, Gelbart WM, Rudnick J. From The Cover: Origin of icosahedral symmetry in viruses. Proc. Natl. Acad. Sci. 2004;101:15556–15560. doi: 10.1073/pnas.0405844101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ritsert K, et al. Studies on the lumazine synthase/riboflavin synthase complex of Bacillus subtilis: crystal structure analysis of reconstituted, icosahedral beta-subunit capsids with bound substrate analogue inhibitor at 2.4 A resolution. J. Mol. Biol. 1995;253:151–167. doi: 10.1006/jmbi.1995.0542. [DOI] [PubMed] [Google Scholar]
- 3.Howorka S. Rationally engineering natural protein assemblies in nanobiotechnology. Curr. Opin. Biotechnol. 2011;22:485–491. doi: 10.1016/j.copbio.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Roldão A, Mellado MCM, Castilho LR, Carrondo MJT, Alves PM. Virus-like particles in vaccine development. Expert Rev. Vaccines. 2010;9:1149–1176. doi: 10.1586/erv.10.115. [DOI] [PubMed] [Google Scholar]
- 5.Effio CL, Hubbuch J. Next generation vaccines and vectors: Designing downstream processes for recombinant protein-based virus-like particles. Biotechnol. J. 2015;10:715–727. doi: 10.1002/biot.201400392. [DOI] [PubMed] [Google Scholar]
- 6.Ma Y, Nolte RJM, Cornelissen JJLM. Virus-based nanocarriers for drug delivery. Adv. Drug Deliv. Rev. 2012;64:811–825. doi: 10.1016/j.addr.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Smith ML, et al. Modified Tobacco mosaic virus particles as scaffolds for display of protein antigens for vaccine applications. Virology. 2006;348:475–488. doi: 10.1016/j.virol.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 8.Bauler P, Huber G, Leyh T, McCammon JA. Channeling by Proximity: The Catalytic Advantages of Active Site Colocalization Using Brownian Dynamics. J. Phys. Chem. Lett. 2010;1:1332–1335. doi: 10.1021/jz1002007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brodin JD, et al. Metal-directed, chemically tunable assembly of one-, two- and three-dimensional crystalline protein arrays. Nat. Chem. 2012;4:375–382. doi: 10.1038/nchem.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Der BS, et al. Metal-mediated affinity and orientation specificity in a computationally designed protein homodimer. J. Am. Chem. Soc. 2012;134:375–385. doi: 10.1021/ja208015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fletcher JM, et al. Self-assembling cages from coiled-coil peptide modules. Science. 2013;340:595–599. doi: 10.1126/science.1233936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Usui K, et al. Nanoscale elongating control of the self-assembled protein filament with the cysteine-introduced building blocks. Protein Sci. Publ. Protein Soc. 2009;18:960–969. doi: 10.1002/pro.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raman S, Machaidze G, Lustig A, Aebi U, Burkhard P. Structure-based design of peptides that self-assemble into regular polyhedral nanoparticles. Nanomedicine Nanotechnol. Biol. Med. 2006;2:95–102. doi: 10.1016/j.nano.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Raman S, et al. Design of Peptide Nanoparticles Using Simple Protein Oligomerization Domains. Open Nanomedicine J. 2009;2:15–26. [Google Scholar]
- 15.Sinclair JC, Davies KM, Vénien-Bryan C, Noble MEM. Generation of protein lattices by fusing proteins with matching rotational symmetry. Nat. Nanotechnol. 2011;6:558–562. doi: 10.1038/nnano.2011.122. [DOI] [PubMed] [Google Scholar]
- 16.Boyle AL, et al. Squaring the circle in peptide assembly: from fibers to discrete nanostructures by de novo design. J. Am. Chem. Soc. 2012;134:15457–15467. doi: 10.1021/ja3053943. [DOI] [PubMed] [Google Scholar]
- 17.Lai Y-T, et al. Structure of a designed protein cage that self-assembles into a highly porous cube. Nat. Chem. 2014;6:1065–1071. doi: 10.1038/nchem.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King NP, et al. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science. 2012;336:1171–1174. doi: 10.1126/science.1219364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King NP, et al. Accurate design of co-assembling multi-component protein nanomaterials. Nature. 2014;510:103–108. doi: 10.1038/nature13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leaver-Fay A, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DiMaio F, Leaver-Fay A, Bradley P, Baker D, André I. Modeling symmetric macromolecular structures in Rosetta3. PloS One. 2011;6:e20450. doi: 10.1371/journal.pone.0020450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 23.Griffiths JS, et al. Cloning, isolation and characterization of the Thermotoga maritima KDPG aldolase. Bioorg. Med. Chem. 2002;10:545–550. doi: 10.1016/s0968-0896(01)00307-8. [DOI] [PubMed] [Google Scholar]
- 24.Fullerton SWB, et al. Mechanism of the Class I KDPG aldolase. Bioorg. Med. Chem. 2006;14:3002–3010. doi: 10.1016/j.bmc.2005.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perlmutter JD, Hagan MF. Mechanisms of Virus Assembly. Annu. Rev. Phys. Chem. 2015;66:217–239. doi: 10.1146/annurev-physchem-040214-121637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pédelacq J-D, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 27.Andrews BT, Schoenfish AR, Roy M, Waldo G, Jennings PA. The Rough Energy Landscape of Superfolder GFP Is Linked to the Chromophore. J. Mol. Biol. 2007;373:476–490. doi: 10.1016/j.jmb.2007.07.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cortese K, Diaspro A, Tacchetti C. Advanced Correlative Light/Electron Microscopy: Current Methods and New Developments Using Tokuyasu Cryosections. J. Histochem. Cytochem. 2009;57:1103–1112. doi: 10.1369/jhc.2009.954214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang P-S, et al. High thermodynamic stability of parametrically designed helical bundles. Science. 2014;346:481–485. doi: 10.1126/science.1257481. [DOI] [PMC free article] [PubMed] [Google Scholar]
References for online-only Methods
- 30.Zhou Z, et al. Genetically encoded short peptide tags for orthogonal protein labeling by Sfp and AcpS phosphopantetheinyl transferases. ACS Chem. Biol. 2007;2:337–346. doi: 10.1021/cb700054k. [DOI] [PubMed] [Google Scholar]
- 31.Baalousha M, Lead JR. Nanoparticle dispersity in toxicology. Nat. Nanotechnol. 2013;8:308–309. doi: 10.1038/nnano.2013.78. [DOI] [PubMed] [Google Scholar]
- 32.Zulauf M, D’Arcy A. Light scattering of proteins as a criterion for crystallization. J. Cryst. Growth. 1992;122:102–106. [Google Scholar]
- 33.Nannenga BL, Iadanza MG, Vollmar BS, Gonen T. In: Current Protocols in Protein Science. Coligan JE, Dunn BM, Speicher DW, Wingfield PT, editors. John Wiley & Sons Inc.; 2013. [Google Scholar]
- 34.Tang G, et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 35.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A New Generation of the IMAGIC Image Processing System. J. Struct. Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 38.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 39.Frank J, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 40.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang P-S, et al. RosettaRemodel: A Generalized Framework for Flexible Backbone Protein Design. PLoS ONE. 2011;6:e24109. doi: 10.1371/journal.pone.0024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller EGD, et al. The organization of the core proteins of the yeast spindle pole body. Mol. Biol. Cell. 2005;16:3341–3352. doi: 10.1091/mbc.E05-03-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimogawa MM, Wargacki MM, Muller EG, Davis TN. Laterally attached kinetochores recruit the checkpoint protein Bub1, but satisfy the spindle checkpoint. Cell Cycle. 2010;9:3619–3628. doi: 10.4161/cc.9.17.12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.