

Abstract
In eukaryotic cells, transcription of every protein-coding gene begins with the assembly of an RNA Polymerase II preinitiation complex (PIC) on the promoter1. The promoters, in conjunction with enhancers, silencers and insulators, define the combinatorial codes that specify gene expression patterns2. Our ability to analyze the control logic encoded in the human genome is currently limited by a lack of accurate information of the promoters for most genes3. Here, we describe a genome-wide map of active promoters in human fibroblast cells, determined by experimentally locating the sites of PIC binding throughout the human genome. This map defines 10,571 active promoters corresponding to 6,763 known genes and at least 1,199 un-annotated transcriptional units. Features of the map suggest extensive usage of multiple promoters by the human genes and widespread clustering of active promoters in the genome. In addition, examination of the genome-wide expression profile reveals four general classes of promoters that define the transcriptome of the cell. These results provide a global view of the functional relationship among the transcriptional machinery, chromatin structure and gene expression in human cells.
The PIC consists of the RNA Polymerase II (RNAP), the transcription factor IID (TFIID) and other general transcription factors4. Our strategy to map the PIC binding sites involves a chromatin immunoprecipitation coupled DNA microarray analysis (ChIP-on-chip), which combines the immunoprecipitation of PIC-bound chromatin from formaldehyde crosslinked cells with parallel identification of the resulting bound DNA sequences using DNA microarrays5,6. Previously, we have demonstrated the feasibility of this strategy by successfully mapping active promoters in 1% of the human genome that correspond to the 44 genomic loci known as the ENCODE regions6,7. To apply this strategy to the entire human genome, we fabricated a series of DNA microarrays8 containing roughly 14.5 million 50-mer oligonucleotides, designed to represent all the non-repeat DNA throughout the human genome at 100 basepairs (bp) resolution. We immunoprecipitated TFIID-bound DNA from the primary fibroblast IMR90 cells with a monoclonal antibody that specifically recognizes the TAF1 subunit of this complex (TBP associated factor 1, formerly TAFII2509, Fig 1a). We then amplified and fluorescently labeled the resulting DNA, and hybridized it to the above microarrays along with a differentially labeled control DNA (Fig. 1a). We determined 9,966 potential TFIID-binding regions using a simple algorithm requiring a stretch of four neighboring probes to have a hybridization signal significantly above the background. To independently verify these TFIID-binding sequences, we designed a condensed array that contained a total of 379,521 oligonucleotides to represent these sequences and 29 control genomic loci selected from the 44 ENCODE regions7 at 100 bp resolution. ChIP-on-chip analysis of two independent samples of IMR90 cells confirmed the binding of TFIID to a total of 8,597 regions, ranging in size from 400 bp to 9.8 Kbp (Fig. 1b). We further defined a total of 12,150 TFIID-binding sites within the 8,597 fragments using a peak finding algorithm that predicts the most likely TFIID-binding sites based on the hybridization intensity of consecutive probes with significant signals (Fig 1b, see supplemental information for details).
Figure 1.
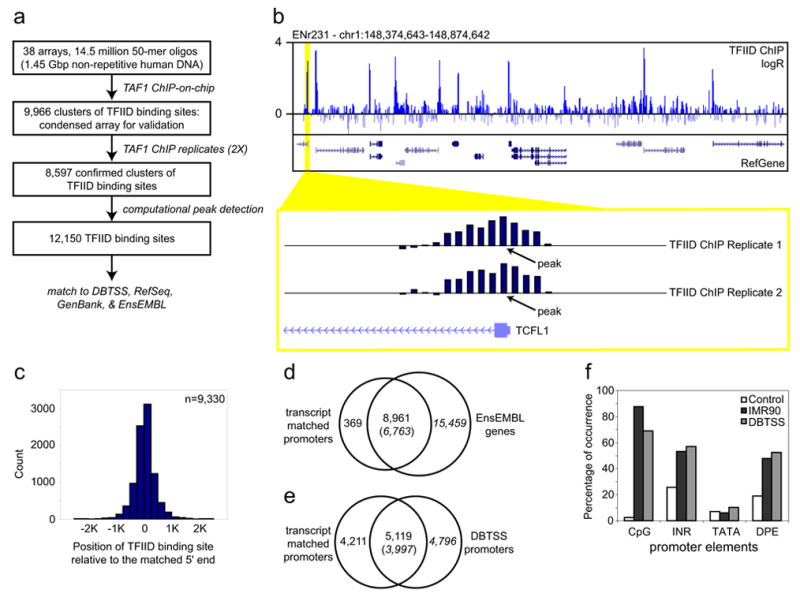
Identification and characterization of active promoters in the human genome. (a) Outline of the strategy employed to map TFIID-binding sites in the genome. (b) A representative view of the results from TFIID ChIP-on-chip analysis. The logarithmic ratio (log2 R) of hybridization intensities between TFIID ChIP DNA and a control DNA, and RefSeq gene annotation is shown in the top and middle panels, respectively. A close-up view of two replicate sets of TFIID ChIP-on-chip hybridization signals around the 5′ end of the TCFL1 gene is shown in the bottom panel. Arrows indicate the position of TFIID-binding site determined by a peak-finding algorithm. (c) Distribution of TFIID-binding sites relative to the 5′ end of the matched transcripts. (d & e) Venn diagrams showing number of identified promoters that matched EnsEMBL genes (d) or promoters annotated in DBTSS (e). (f) Chart showing the percentage of IMR90 or DBTSS promoters overlapping with CpG islands, or containing conserved TATA box, INR or DPE elements (see supplemental information for details).
Next, we matched these 12,150 TFIID-binding sites to the 5′ end of known transcripts in three public transcript databases (DBTSS10, RefSeq11, GenBank human mRNA collection12) and the EnsEMBL gene catalog13. To account for the uncertainty of our knowledge of the true 5′ end of transcripts and the uncertainty of predicted TFIID-binding positions due to noise within the microarray data, we chose an arbitrary distance of 2.5 Kbp as a measure of close proximity. We found that 10,553 (87%) TFIID-binding sites were within 2.5Kbp of annotated 5′ ends of known mRNA. We resolved common TFIID-binding sites mapping to similar 5′ ends to define a non-redundant set of 9,330 5′ end-matched TFIID-binding sites. Of these TFIID-binding sequences 7,789 (83%) were found within 500 bp of the putative transcription start sites (TSS) (Fig. 1c). Since these 9,330 DNA sequences were bound by TFIID in vivo and within close proximity to the 5′end of known transcripts, we defined them as promoters for the corresponding transcripts (Table S1). Of these 9,330 promoters, 8,961 were mapped within 2.5 Kbp of the 5′ end or within annotated boundaries of 6,763 known genes in the EnsEMBL gene catalog13 (Fig. 1d, Table S1). The remaining 369 promoters corresponded to transcripts not contained within these boundaries of EnsEMBL genes, and therefore provide support for inclusion of these transcripts to the current gene catalogs. The list of promoters also confirmed 5,119 previously annotated promoters10, and defined 4,211 new promoters for at least 2,627 genes (Fig. 1e, Table S1).
Four independent analyses validated the high specificity and accuracy of the active promoters detected in IMR90 cells. First, ChIP-on-chip analysis using an anti-RNAP antibody (8WG16) confirmed the binding of RNAP to at least 9,050 (97%) of the 9,330 promoters in IMR90 cells (Fig. S1). Second, standard chromatin immunoprecipitation (ChIP) performed on 28 promoters randomly selected from the above list confirmed the occupancy of RNAP on all but one promoter (Fig. S2). Third, the 9,330 active promoters are enriched for known promoter-associated sequences such as CpG islands, and the INR and DPE core promoter elements (Fig. 1f). The percentage of CpG-associated promoters (88%) was significantly higher than the previous estimate (56%)14, suggesting that CpG islands may play a more general role in gene expression than previously appreciated. Surprisingly, we did not find the TATA box to be significantly enriched in these promoters (Fig. 1f). This may be due to a lack of conservation of the TATA box in human promoters; alternatively, this may indicate that the TATA box is not a general promoter motif for human genes. This observation is in line with previous reports that the TATA box is only present in a small number of promoters in yeast and in Drosophila15. Fourth, ChIP-on-chip analysis using antibodies that recognize acetylated histone H3 (AcH3) or di-methylated lysine 4 on histone H3 (MeH3K4) showed that over 97% of the 9,330 promoters were associated with these known epigenetic marks for active genes (Fig. 2a)16. Interestingly, the localization of MeH3K4 in these promoters was predominantly downstream of the TFIID-binding site (Fig. 2b), and the mechanisms for such chromatin organization at human promoters are currently unknown.
Figure 2.
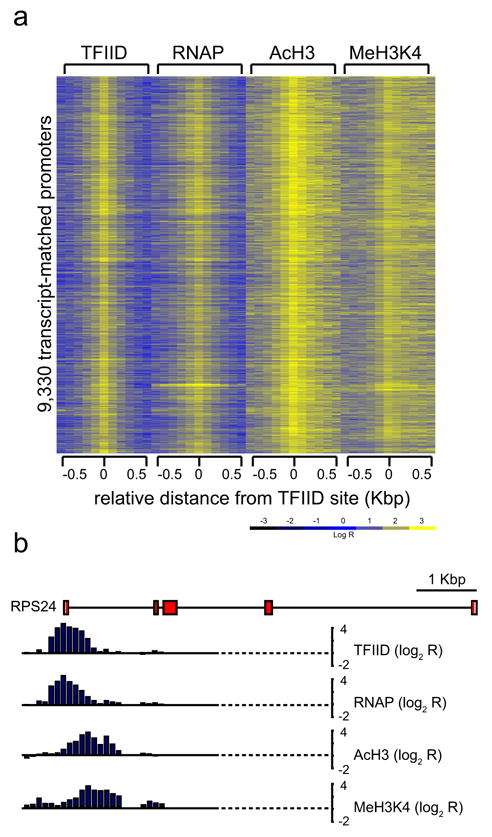
Chromatin modification features of the active promoters. Logarithmic ratios of the ChIP-on-chip hybridization intensities (log2 R) of probes from 0.5 Kbp upstream to 0.5 Kbp downstream of the identified TFIID-binding sites for TFIID, RNAP, AcH3, and MeH3K4 are plotted in a yellow-blue colored scale for 9,330 transcript-matched promoters. The bottom panel shows a yellow-blue colored scale used to color each cell with corresponding log2 R values. (b) A detailed view of TFIID, RNAP, AcH3, and MeH3K4 profiles on the promoter of RPS24 gene.
Among the 12,150 mapped TFIID-binding sites, 1,597 are found more than 2.5 Kbp away from previously defined 5′ ends of mRNA, and may represent promoters for novel transcripts or genes (Table S2). Of these, 607 non-redundant TFIID-binding sites were matched within 2.5 Kbp of the 5′ ends of the Expressed Sequence Tag (EST)-based gene models, indicating that they may indeed produce mRNA (Table S2). The remaining TFIID-binding sites were further filtered to a set of 634 putative promoters by requiring the occupancy of RNAP and presence of AcH3 and MeH3K4 within 1 Kbp of these sites (Fig. S3). To verify that these promoters drive transcription, we analyzed mRNA from the IMR90 cells, using 50-mer oligonucleotide arrays that represent a 28 Kbp sequence surrounding 569 of 634 unmatched putative promoters. At least 36 novel transcription units were identified near the putative promoter regions, suggesting that these may represent new transcription units yet to be annotated in the human genome (Table S3). The failure to detect mRNA from the other putative promoters may indicate that these transcripts are highly unstable. Indeed, at least one putative promoter is located within 250 bp upstream from a predicted miRNA17 (Fig. S4), suggesting that some putative promoters could transcribe non-coding RNA that might have escaped detection by conventional mRNA isolation techniques.
In all, we defined a set of 1,241 putative promoters that correspond to previously un-annotated transcription units (Fig. 4b, Table S2). Evolutionarily conserved regions were found in a majority of these putative promoters (Fig. S5). In addition, they were significantly enriched for core promoter motifs including INR (46%) and DPE (40%) and overlapped with CpG islands (40%, Fig. S6). These results suggest that many of the putative promoter sequences that we have defined by TFIID-binding sites may indeed be functional promoters. There are 830 putative promoters located in the intergenic regions. These promoters, together with the 369 promoters that matched to transcripts outside the EnsEMBL genes, may suggest the existence of 1,199 novel transcription units outside the current gene annotation18. This number corresponds to about 13% of the 8,961 promoters that were matched to known genes; therefore, we estimate that there are likely additional 13% of the human genes that remain to be annotated in the genome. This number agrees well with a recent estimate of the total number of human genes18, but is considerably lower than estimates based on number of transcripts detected by microarrays, SAGE, and other methods19–22. It is conceivable that promoters for many low-abundance transcripts may be infrequently occupied by TFIID and possibly escaped detection by our assays. Alternatively, it is possible that the novel transcripts detected by the other studies are products from a different transcription machinery or process.
Figure 4.
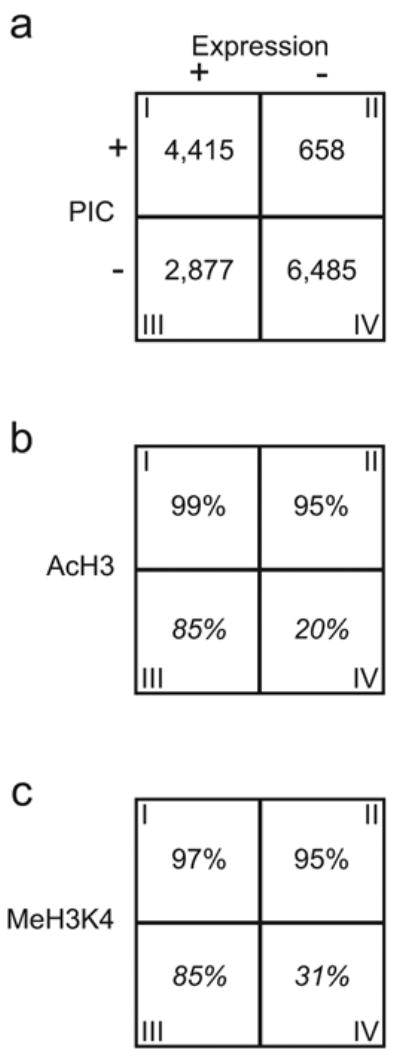
Four distinct classes of promoters define the transcriptome of IMR90 cells. (a) A 2x2 matrix describes the distribution of genes defined by expression and PIC occupancy on the promoter. (b & c) Matrices showing the percentages of genes associated with the AcH3 (b) or MeH3K4 (c) modification for each of the four classes of genes. Italicized numbers in some boxes represent extrapolation from the 29 ENCODE regions.
Two notable features were apparent in this map of active promoters. First, large domains of four or more consecutive genes were found to be simultaneously bound by PIC and likely transcribed in the IMR90 cells. At least 256 clusters, consisting of 1,668 EnsEMBL genes, can be classified into such regions, and the number of clustered promoters is highly significant (P ≪ 0.001, Table S5). The clustering of active promoters is consistent with previous findings that co-regulated genes tend to be organized into coordinately regulated domains23–26. Second, a large number of genes contained two or more active promoters (Table S4). In general, these multiple promoters correspond to transcripts with either different 5′ UTR sequences or distinct first exons (i., PTEN) but do not affect the open reading frames. In some cases, however, distinct proteins were produced from multiple promoters (i., NR2F2, WEE1). In other cases, transcripts undergo differential splicing and polyadenylation (i., NFKB2, STAT3). The widespread usage of multiple promoters in this single cell type indicates a greater complexity of the cellular proteome than previously expected and also reveals highly coordinated regulation of transcriptional initiation, splicing, and polyadenylation throughout the genome27. To experimentally verify our observations regarding multiple promoter utilization in IMR90 cells, we selected the WEE1 gene for further analysis. Two TFIID-binding sites were mapped within this gene, corresponding to the 5′ ends of two distinct mRNAs, NM_003390 and AK122837 (Fig. 3a). Each mRNA encodes a distinct protein: one encodes a well-characterized full length version of WEE1 protein, and the other only the kinase domain. We detected both transcripts in a steady state, asynchronous population of IMR90 cells (Fig. 3b). Interestingly, the shorter transcript appears to be most abundant in G0 phase, while the longer transcript is highly transcribed in both G0 and S phase (Fig. 3c), suggesting that the two promoters in the WEE1 gene may have distinct cell cycle functions.
Figure 3.

Utilization of multiple promoters by human genes. (a) Annotation of the WEE1 gene locus and the corresponding TFIID-binding profile. Black bars over the first and second exons in transcripts indicate the positions of the primers used for real-time quantitative RT-PCR analysis of each transcript. (b) RT-PCR analysis of NM_003390 and AK122837 transcripts in asynchronous population of IMR90 cells. (c) Real-time quantitative RT-PCR analysis of NM_003390 and AK122837 transcripts in cell cycle synchronized population of IMR90 cells. Transcript levels observed for each cell cycle phase were normalized to the level observed in the asynchronous population.
The active promoter map in IMR90 cells allowed us to systematically investigate the functional relationship between the transcription machinery and gene expression. We examined the genome-wide expression profiles of IMR90 cells and correlated the expression status of 14,437 EnsEMBL genes to promoter occupancy by the PIC. The comparison revealed four general classes of genes (Fig. 4, Table S6). Class I consists of 4,415 genes whose promoters were bound by the PIC, and transcripts were detected. Class II includes 658 genes whose promoters were bound by the PIC, but no transcript was detected. Class III contains 2,879 genes that were transcribed in IMR90 cells but the PIC was not detected on their promoters. Class IV comprises of the remaining 6,485 genes whose promoters were not bound by PIC and their corresponding transcripts were not detected.
The genes in class I and class IV, representing over 75% of the genes examined, support the general model that formation of the PIC on the promoters leads to transcription. The class II and III genes, on the other hand, are inconsistent with this model and may indicate other mechanism is responsible for expression of these genes. We postulate that the discrepancy between the PIC formation and transcription on the class II promoters are due to at least two possibilities. The first possibility is that the PIC assembles on these promoters, but the PIC formation is not sufficient to initiate transcription. Additional regulatory steps, such as promoter clearance or elongation may be rate-limiting in transcription of these genes28. Some notable examples in class II are the immediate early genes, FOS and FOSB; the heat shock protein genes, HSPA6 and HSPD1; and the DNA damage repair genes, MSH5 and ERCC4. The second possibility is that transcription actually takes place at these promoters, but the resulting mRNAs are post-transcriptionally degraded, as in miRNA-mediated post-transcriptional silencing29.
In contrast to class II, genes in class III appear to be transcribed, but the PIC binding on their promoters was not detected. This could simply be due to moderate sensitivity of our method6. To address this issue, we performed standard ChIP assay to detect binding of TFIID and RNAP on 10 randomly selected class III gene promoters. Nearly 60% of the promoters were weakly associated with TFIID and RNAP in these cells, and were marked by enrichment ratios less than 2-fold but nonetheless above the observed background (Fig. S2). Hence, the failure to detect TFIID and RNAP occupancy in roughly 60% of the class III promoters (~1,700) may be due to weak signals that fall below the detection sensitivity of our method. This result indicates that the promoters of a significant fraction of class III genes are open and accessible for transcription, but PIC assembles on these promoters transiently, weakly or only during the early stage of fibroblast differentiation.
In order to understand the functional relationship between the histone modification status and gene expression, we examined the histone modifications (AcH3 and MeH3K4) in 29 ENCODE regions7 (Table S7), with a specific focus on the four classes of gene promoters. As expected, these epigenetic markers were associated with virtually all class I and class II genes, and the vast majority of class III genes. However, roughly 20% of the class IV genes were also associated with these markers (Fig. 4). This result suggests that a significant number of genes not actively transcribed are also associated with these epigenetic markers. We speculate that these histone modifications may serve to restrict genome expression potential and define the transcriptome capacity of the cell, and the transcription regulators and machinery collaborate with these epigenetic markers to further restrict the transcriptome to generate a unique pattern of genome expression.
Our results provide an initial framework for analysis of the cis-regulatory logic30 in human cells. The high-resolution map of active promoters in IMR90 cells will enable detailed analysis of transcription factor binding sites within these regions. The promoter map described here can also serve as a reference to understand gene expression in other cell types. We expect that a survey of additional cell types using the same approach will allow comprehensive mapping of all promoters in the human genome, and help elucidate the control logic that governs gene expression in different cell types in the body.
Methods
Supplementary Material
Acknowledgments
We thank Jim Kadonaga, Richard A. Young, Richard Kolodner, Webster K. Cavenee, Sara Van Calcar, and Christopher K. Glass for discussion and their comments on the manuscript. This research was supported by Ruth L. Kirschstein National Research Service Award F32CA108313 (T. H. K.); Ford Foundation Predoctoral Fellowship (L. O. B.); Ludwig Institute for Cancer Research (B. R.); U01HG003151 (B. R.) and R21CA105829 (B. R.) from NIH; and IIS-0222967 from NSF (Y. W.).
References
- 1.Smale ST, Kadonaga JT. The RNA polymerase II core promoter. Annu Rev Biochem. 2003;72:449–79. doi: 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- 2.Tjian R, Maniatis T. Transcriptional activation: a complex puzzle with few easy pieces. Cell. 1994;77:5–8. doi: 10.1016/0092-8674(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 3.Trinklein ND, Aldred SJ, Saldanha AJ, Myers RM. Identification and functional analysis of human transcriptional promoters. Genome Res. 2003;13:308–12. doi: 10.1101/gr.794803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reinberg D, et al. The RNA polymerase II general transcription factors: past, present, and future. Cold Spring Harb Symp Quant Biol. 1998;63:83–103. doi: 10.1101/sqb.1998.63.83. [DOI] [PubMed] [Google Scholar]
- 5.Ren B, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–9. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 6.Kim TH, et al. Direct isolation and identification of promoters in the human genome. Genome Res. 2005;15 doi: 10.1101/gr.3430605. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–40. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 8.Singh-Gasson S, et al. Maskless fabrication of light-directed oligonucleotide microarrays using a digital micromirror array. Nat Biotechnol. 1999;17:974–8. doi: 10.1038/13664. [DOI] [PubMed] [Google Scholar]
- 9.Ruppert S, Wang EH, Tjian R. Cloning and expression of human TAFII250: a TBP-associated factor implicated in cell-cycle regulation. Nature. 1993;362:175–9. doi: 10.1038/362175a0. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki Y, Yamashita R, Sugano S, Nakai K. DBTSS, DataBase of Transcriptional Start Sites: progress report 2004. Nucleic Acids Res. 2004;32(Database issue):D78–81. doi: 10.1093/nar/gkh076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence project: update and current status. Nucleic Acids Res. 2003;31:34–7. doi: 10.1093/nar/gkg111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank: update. Nucleic Acids Res. 2004;32(Database issue):D23–6. doi: 10.1093/nar/gkh045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birney E, et al. Ensembl 2004. Nucleic Acids Res. 2004;32(Database issue):D468–70. doi: 10.1093/nar/gkh038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antequera F, Bird A. Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci U S A. 1993;90:11995–9. doi: 10.1073/pnas.90.24.11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohler U, Liao GC, Niemann H, Rubin GM. Computational analysis of core promoters in the Drosophila genome. Genome Biol. 2002;3:RESEARCH0087. doi: 10.1186/gb-2002-3-12-research0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schubeler D, et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004;18:1263–71. doi: 10.1101/gad.1198204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32(Database issue):D109–11. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 19.Bertone P, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–6. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- 20.Kampa D, et al. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 2004;14:331–42. doi: 10.1101/gr.2094104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saha S, et al. Using the transcriptome to annotate the genome. Nat Biotechnol. 2002;20:508–12. doi: 10.1038/nbt0502-508. [DOI] [PubMed] [Google Scholar]
- 22.Rinn JL, et al. The transcriptional activity of human Chromosome 22. Genes Dev. 2003;17:529–40. doi: 10.1101/gad.1055203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su AI, et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A. 2002;99:4465–70. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spellman PT, Rubin GM. Evidence for large domains of similarly expressed genes in the Drosophila genome. J Biol. 2002;1:5. doi: 10.1186/1475-4924-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roy PJ, Stuart JM, Lund J, Kim SK. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature. 2002;418:975–9. doi: 10.1038/nature01012. [DOI] [PubMed] [Google Scholar]
- 26.Caron H, et al. The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science. 2001;291:1289–92. doi: 10.1126/science.1056794. [DOI] [PubMed] [Google Scholar]
- 27.Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature. 2002;416:499–506. doi: 10.1038/416499a. [DOI] [PubMed] [Google Scholar]
- 28.Krumm A, Hickey LB, Groudine M. Promoter-proximal pausing of RNA polymerase II defines a general rate-limiting step after transcription initiation. Genes Dev. 1995;9:559–72. doi: 10.1101/gad.9.5.559. [DOI] [PubMed] [Google Scholar]
- 29.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 30.Yuh CH, Bolouri H, Davidson EH. Genomic cis-regulatory logic: experimental and computational analysis of a sea urchin gene. Science. 1998;279:1896–902. doi: 10.1126/science.279.5358.1896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.