

SUMMARY
Metastasis is the primary cause of cancer-related deaths. While The Cancer Genome Atlas (TCGA) has sequenced primary tumor types obtained from surgical resections, much less comprehensive molecular analysis is available from clinically acquired metastatic cancers. Here, we perform whole exome and transcriptome sequencing of 500 adult patients with metastatic solid tumors of diverse lineage and biopsy site. The most prevalent genes somatically altered in metastatic cancer included TP53, CDKN2A, PTEN, PIK3CA, and RB1. Putative pathogenic germline variants were present in 12.2% of cases of which 75% were related to defects in DNA repair. RNA sequencing complemented DNA sequencing for the identification of gene fusions, pathway activation, and immune profiling. Integrative sequence analysis provides a clinically relevant, multi-dimensional view of the complex molecular landscape and microenvironment of metastatic cancers.
Keywords: precision medicine, clinical sequencing, metastatic cancers, whole exome sequencing, transcriptome sequencing
Introduction
Tumor metastasis is the process in which cancer cells disperse from a primary site and progressively colonize distant organs. In over 90% of cases, metastatic spread of tumor cells is the greatest contributor to deaths from cancer1,2. With the preponderance of cancer patients enrolled in early stage (Phase I-II) clinical trials harboring metastatic disease2, and with the advent of genomic testing of tumors, there remains the promise of matching patients to the right therapy based on comprehensive molecular profiles3 of pathogenic somatic4,5 and germline6 variants, and components of the functional genome, tumor phenotype, and tumor microenvironment afforded by RNA sequencing7,8. While metastatic tumors share key driver mutations with the primary tumor from which they arose, they often develop new mutations as they evolve during metastasis and treatment9. Thus, it is preferable to match patients to potential therapies and clinical trials based on a real-time analysis of their metastatic tumor, rather than archival material of their primary tumor2,10.
In 2010, we introduced the Michigan Oncology Sequencing (Mi-Oncoseq) Program, an IRB-approved protocol to carry out prospective, integrative exome and transcriptome sequencing of advanced cancer patients7, mirroring the efforts of the TCGA project which focused on generating exome and transcriptome sequence11 in primary cancers. It was among the first comprehensive, clinical DNA and RNA sequencing programs offered for cancer patients8,12–16. The purpose of the Mi-Oncoseq program was to determine the utility of genomic sequencing of tumors and germline coupled with a multi-disciplinary Precision Medicine Tumor Board (PMTB) in the management of advanced cancer patients7,15. The program transitioned into sequencing in a clinical setting (under CLIA) as part of the Clinical Sequencing Exploratory Research (CSER) consortium in 201314–16.
In this study, we carried out clinical grade whole exome (tumor/normal) and transcriptome sequencing (i.e., integrative sequencing) of 500 cancer patients harboring metastatic cancers from over 30 primary sites and biopsied from over 22 organs (abbreviated as the “MET500” cohort). Sequencing matched tumor and normal samples from patients delineated potentially pathogenic germline alterations as well as provided high resolution copy number landscapes. RNA sequencing analysis provided insights into functional gene fusions, transcriptional pathway activation, and a landscape of immune infiltration.
The landscape of molecular aberrations in metastatic cancers
We successfully obtained 537 biopsies from 556 enrolled patients and obtained complete sequencing results on 500 patients with metastatic cancers, representing a 93% success rate. Reasons for failure included lack of tumor content on biopsy (37 cases, 6.6%), biopsy material not available (19 cases, 3.4%; patient declined biopsy, poor physical performance, unable to image site, unsafe for biopsy, insufficient tissue, or enrolled in other clinical trials). The majority of patients, 468 (93.6%) were patients seen at the University of Michigan Comprehensive Cancer Center, however patients from 21 other institutions were also enrolled. The patient demographics were 258 (51.6%) males, 242 females (48.4%), 460 (92%) Caucasian, and 40 (8%) non-Caucasian. The median age of the cohort was 59, with a range of 18 to 86 (Supplementary Table 1 and Extended Data Fig. 1a, b).
Fig. 1a portrays the cancer types (n=20) represented in the MET500 cohort. The top three cancer types in our cohort include 93 (18.6%) metastatic prostate cancers, 91 (18.2%) metastatic breast cancers, and 42 (8.4%) soft tissue sarcomas. There were also 25 (5%) carcinomas of unknown primary (CUP). Fig. 1b highlights the diverse metastatic sites analyzed (n>30) in the MET500 cohort. The most prevalent sites of metastases included 134 liver, 114 lymph node, 46 lung, 42 bone, and 32 abdominal mass/ascites/pleural fluid.
Figure 1. Landscape of molecular alterations in metastatic cancer.

a. Cancer types in the MET500 cohort. Number of cases indicated for each cancer type. b. Site of biopsies. c Mutational burden across tumor types from the MET500 and corresponding primary TCGA cohorts. Transparent boxplots signify insignificant differences (Wilcoxon rank-sum test FDR >= 0.1) d and e. Landscape of molecular alterations in the MET500 cohort. Each cell represents the mutation status of an individual gene for a select patient. Putative oncogenes are represented on panel d and putative tumor suppressor genes in panel e. The percentage of mutations across the MET500 cohort is represented by vertical histograms.
For each patient we performed paired exome sequencing on tumor and germline DNA in order to identify likely pathogenic variants and resolve their somatic or germline origin. Mean target coverages for tumor and normal exomes were 180X and 120X. The average tumor content was 62%. Sequencing metrics are summarized in Supplementary Table 2. Within the targeted regions, we identified an average of 119 somatic mutations per patient. For the majority of cancer types the number of mutations significantly increased in metastases relative to primary tumors in TCGA (Fig. 1c). The difference was more pronounced for tumor types with low mutation burden in the primary stage, e.g. prostate or adrenal cancer. To identify the most recurrent, and hence likely pathogenic, targets of genetic alterations, we performed an integrative analysis of single nucleotide variants (SNV), copy-number variants (CNV), and gene fusions. For each patient and gene, we classified the most recurrently mutated genes as putative tumor-suppressors or oncogenes based on the increase in frequency of inactivating mutations and expert knowledge (Fig. 1d and e, Supplementary Table 3). We found a long-tailed mutational spectrum for both tumor-suppressors and oncogenes. TP53 (266, 53.2%), CDKN2A (80, 16%), PTEN (79, 15.8%) and RB1 (68, 13.6%), were the most frequently altered tumor suppressors, while the most frequently mutated oncogenes included PIK3CA (67, 13.4%), AR (63, 12.6%) and KRAS (51, 10.2%). Overall, tumor suppressors were altered across many cancer types (e.g., TP53 and RB1), while oncogenes were more strongly associated with individual cancer types (e.g., AR or GNAS) (Extended Data Fig. 1c). We further compared the alteration frequencies to those from primary tumors and found that the increase in mutation burden (Fig. 1c) was mirrored by an increase in the frequency of genetic aberrations for the most widely mutated genes (Extended Data Fig. 1d).
Germline variants in metastatic cancer
Through sequencing matched germline DNA, we identified 63 presumed pathogenic germline mutations (PPGM), as defined by ClinVar expert curation, involving 18 genes in 61 individuals (12.2%) (Fig. 2a). These included 30 deleterious missense mutations, 8 nonsense mutations, 20 frameshift mutations, and 5 deleterious splice site mutations (Fig. 2b and Supplementary Table 4). Seventy-five percent of the PPGMs were in genes related to DNA repair with MUTYH (n=10, 16%), BRCA2 (n=9, 14%), CHEK2 (n=9, 14%), and BRCA1 (n=5, 8%) the most common. Outside of DNA repair pathways, we observed PPGMs in APC (n=6, 9.5%), MITF (n=5, 8%), and HOXB13 (n=3, 5%), among other genes. Of the 63 instances of pathogenic alleles identified, 5 alleles were previously unreported, while the remaining alleles have existing ClinVar entries with assigned pathogenic or likely pathogenic significance. Of the 61 individuals with PPGMs identified in this study, 30 (49%) had a somatic second allele aberration within the tumor genome, including LOH and exhibited molecular phenotypes consistent with pathogenicity (Supplementary Table 4). The remaining cases were of carrier status for the identified allele.
Figure 2. Putative pathogenic germline variants in metastatic cancers.

a. Pathogenic germline alterations identified in the MET500 cohort. DNA repair pathway related variants are indicated in shades of blue while “other” alterations are indicated in shades of red. b. Gene level schematic of pathogenic germline variants identified in the MET500 cohort.
Next, we compared the frequencies in genes with PPGMs discovered in the MET500 cohort to the population frequencies in 52,790 individuals compiled by the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/) excluding TCGA cancer samples17. The odds of any PPGM in metastatic cancer significantly exceeded the odds found in the populations comprising ExAC (OR = 3.00, 2.28–3.9, P=1 × 10−13). The genes analyzed and found to be enriched in the metastatic series included BRCA1, BRCA2, APC, CHEK2, MITF, MLH1, NBN and RB1 (Supplementary Table 5).
The gene fusion landscape of metastatic cancer
To identify both activating and inactivating gene fusions we analyzed 868 transcriptome libraries from 496 metastatic tumor RNAs. These fusion junctions involved 12,027 unique gene pairs, an average of 34 gene fusions per tumor, derived from a range of structural aberrations (Fig. 3a). Large differences in fusion-burden were observed across tumor type (Extended Data Fig. 2a). 199 cases (39.8%) harbored at least one putative pathogenic fusion with 138 activating fusions and 103 deleterious fusions (Supplementary Table 6). The activating fusions could be classified as DNA-binding (n=88), protein kinases (n=29), and signal transducers (n=21) (Fig. 3b). The loss-of-function fusions segregated into canonical tumor suppressor genes (n=59), chromatin modifying genes (n=35), and genes involved in cell adhesion (n=9). The most commonly fused tumor suppressor genes were NF1 (n=18), TP53 (n=11), PTEN (n=11), and RB1 (n=6) (Extended Data Fig. 2b). Interestingly, we identified a series of 8 novel fusion pairs in metastatic cancers that we believe are pathogenic (Fig. 3c). These include activated FGFR, BRAF, and ALK fusions with novel partners, extending the range of both fusion partners and cancer types for these clinically targetable fusions.18–22 Novel gene fusions with functional domains include GREB1-NR4A3 in uterine leiomyosarcoma, POC5-PRKD1 in polymorphous low-grade adenocarcinoma of the tongue, and CIC-CITED1 in undifferentiated high-grade sarcoma. Notch fusions fall into 2 classes, those predicted to be sensitive to gamma-secretase inhibition (e.g., NOTCH2-SPAG17) and fusions which are independent of gamma-secretase processing (e.g., PARS2-NOTCH2).
Figure 3. Diverse classes of gene fusions identified in metastatic cancers.

a. Fusions classified by underlying structural aberrations. b. Functional gene fusions identified in the MET500 cohort. c. Molecular structure of novel, potentially activating, gene fusions in the MET500 cohort..
Transcriptional signatures of metastatic disease
To investigate the potential clinical utility of metastatic expression profiles, we analyzed transcriptomes of the 496 biopsy samples (868 libraries). We first evaluated to what extent tissue and cancer specific gene expression is maintained across metastatic lesions. We used the t-SNE projection23 to qualitatively visualize the expression of primary cancer marker across the MET500. Compared to primary tumors, metastatic samples were less well separated, more heterogeneous, and did not segregate based on biopsy site, with the exception of liver biopsies (Fig. 4a, Extended Data Fig. 2c–d),. We compared the expression of tissue-specific marker genes derived from 36 normal tissues24 between normal, primary, and metastatic samples, and observed significant dedifferentiation with disease progression (Extended Data Fig. 2e).
Figure 4. Diverse transcriptional profiles of metastatic cancers.

a. Global gene expression patterns of the MET500 identify poorly differentiated cancers as illustrated by a t-SNE projection of the MET500 samples. Position of samples within the plot reflects the relative similarity in the expression of cancer-specific markers. Samples are color-coded based on their assigned analysis cohort. b. Correlation heatmap and hierarchical clustering showing similarities (red) and dissimilarities (blue) in the transcriptional activity of computationally derived aggregate sets of signatures across the MET500 “meta-signatures”. c. Negative correlation between signatures of EMT and proliferation (S-phase of the cell cycle, FANCA pathway). All MET500 samples are shown.
Next, we looked at transcriptional signatures associated with perturbed cancer-related genes25,26. Compared to normal tissues, transcriptional output was increased for most oncogenic signatures (Extended Data Fig. 3a–b), indicating a global shift towards a cancer-related transcriptional program. Unsupervised clustering of signature scores across patients revealed relevant associations between gene sets, and phenotypic similarities among patients (Extended Data Fig. 4). Inference of patient-specific activities27,28 revealed coordinated changes across curated pathways that coalesce into a small number of principal cancer hallmarks (Extended Data Fig. 5a–b): interferon response, inflammatory response, epithelial to mesenchymal transition (EMT), proliferation, and metabolism. Importantly, these associations were robust to algorithm choice. Compared to normal tissues, metastatic tumors show a global increase in proliferation, stress-response, and metabolism. Conversely, hallmarks of EMT and cancer-immune responses can be either up- or down-regulated (Extended Data Fig. 5a–b). Next, we computationally delineated 25 non-redundant experimental “meta-signatures” (Extended Data Fig. 6). Unsupervised clustering and correlation analysis of meta-signatures revealed four of the canonical cancer hallmarks: immune response, EMT, proliferation, and metabolism (Fig. 4b). Metastatic tumors fall into two main subtypes: an EMT-like subtype associated with inflammation signatures29, and a proliferative subtype associated with increased metabolism and systemic stress. In agreement, we observed mutual exclusivity between curated proliferative and EMT gene sets (Fig. 4c). Interestingly, this trend was less prominent across primary tumors (Extended Data Fig. 7a). Importantly, meta-signature activities were found to be weakly associated with biopsy site (Extended Data Fig. 7b) and primary tissue (Extended Data Fig. 7c), and held independently for common cancer types and biopsy sites (Extended Data Fig. 7d).
The immune microenvironment of metastatic disease
To characterize the phenotype of host-immune responses, we leveraged exome, RNA-seq, and a dedicated assay for T-cell repertoire profiling. Based on immune-cell markers proposed by Yoshihara et al.30, we developed an RNA-seq based score, MImmScore, to assess the magnitude of leukocyte infiltration. We found that MImmScores is negatively correlated with tumor content (Extended Data Fig. 8a), and positively correlated with stromal infiltration (Extended Data Fig. 8b). MImmScores were compared to canonical T-cell expression markers (RNA-seq based) and DNA-based T-cell receptor β CDR3 sequencing, and all three measures are in good agreement (Extended Data Fig. 8c–d). We also discovered that metastatic immune infiltration is strongly determined by tumor type (Fig. 5a) and to a lesser degree by biopsy site (Extended Data Fig. 9a). Cancer types known to be infiltrated in the localized stage (Extended Data Fig. 9b), including kidney cancer31,32, lung cancer33, and melanoma34, remained infiltrated at metastatic sites. Less immunogenic types, such as breast and prostate cancer, were generally associated with lower MImmScores in the primary and metastatic stage (Fig. 5a, Extended Data Fig. 9b). Immune infiltration was found to be heterogeneous not only across cancer types, but also within individual cohorts (Extended Data Fig. 9c–d). Strikingly, individual patients with high levels of immune infiltration could be identified even within tumor types that did not thus far respond to immunotherapies.
Figure 5. The immune microenvironment of metastatic cancers.

a. Magnitude of immune (leukocyte) infiltration (MImmScore) across the MET500 analysis cohort. b. Hierarchical clustering of samples by their predicted immune infiltrates. c–d. T-cell receptor profiling by TCRβ DNA deep-sequencing. Correlation of estimated T-cell numbers (templates): c. with clonal expansion (high clonality indicates that many T-cells have the same TCRβ sequence) (rho - Spearman’s rank-correlation coefficient). d. with number of mutations. e. Clusters of patients based on the normalized expression levels of APC (left) or T-cell (right) surface molecules. f–h. genomic correlates for patients grouped by their membership in immunologically active clusters: TIL-5, APC-1, Tcell-1 (silent=none, partial=some, complete=all). f. expression of PD-L1 (t-test). g. number of non-synonymous mutations (Wilcoxon test). h. response-score based on a predictive gene expression signature to immunotherapy (t-test). Significance levels: * (p=0.05–0.001), ** (p=0.001–1e-6), *** (p=<1e-6).
We hypothesized that metastatic tumors differ not only in the magnitude but also composition, in terms of leukocyte cell types, of tumor infiltrating lymphocytes (TILs) and macrophages (TAMs). Unsupervised clustering revealed groups of samples with significant differences in TIL composition, based on bulk tumor transcriptome data35 (Fig. 5b). Cancers were most strongly typified by the different ratios of M2 to M0 (unpolarized) macrophages (clusters TIL-2,4,6,7) and different CD8+ to CD4+ T-cell ratios (high CD8+ TIL-1, high CD4+ TIL-4). While immunosuppressive M2 macrophages36 were highly prevalent, pro-inflammatory, anti-tumor M1 macrophages were largely absent. A small cluster of samples (TIL-5) was characterized by a dominant ratio of cytotoxic CD8+ T-cells. To assess clonal T-cell expansion, we selected index cases with a high MImmScore and CD8+ T-cell ratio or with low immune infiltration. We ascertained the identity and frequency of T-cell clones by T-cell receptor β (CDR3) deep-sequencing, and found that the estimated numbers of T-cells were dramatically increased in the index cases (Supplementary Table 7). Most importantly, this increase was correlated with a significant expansion of T-cell clones (increased clonality) (Fig. 5c, Extended Data Fig. 9e) and a concomitant decrease in the ratio of Tregs to cytotoxic T-cells (Extended Data Fig. 9f). Highly mutated samples were found to be associated with a larger number of infiltrating T-cells (Fig. 5d) and increased MImmScores (Extended Data Fig. 10a).
Next, we focused on the expression of ligand /receptor pairs on the surface of T-cells and APCs. These molecules are either co-stimulatory and required for T-cell activation, or co-inhibitory as in the case of immune checkpoints (Fig. 5e). The majority of patients were either immunologically silent (clusters Tcell-0, APC-0), or immunologically active (Tcell-1, APC-1), with a highly significant overlap between the independent cluster analyses (Extended Data Fig. 10b–c). Importantly, almost all samples in APC-1 express CD80/CD86 and almost all samples in Tcell-1 express CD28. CD80/CD86 are the ligands for the CD28 receptor and a critical signal for T-cell activation.
Finally, we examined the relationships between the emerging predictive biomarkers for immune therapy and the transcriptomic immune phenotypes. We stratified patients into three categories: immunologically silent, partially active, and fully active. A sample was categorized as completely or partially active if it was a member of all or at least one of the active clusters: TIL-5, APC-1, Tcell-1, respectively. Patients in the active categories exhibited increased levels of expression biomarkers: PD-L137 (Fig. 5f), HLA38, and granzyme39 and had higher mutational burden (Fig. 5g), which is both a prognostic and predictive marker40. Finally, leveraging a predictive signature to immunotherapy in metastatic melanoma41, we developed a clinical-response score. As expected, immunologically active patients had significantly higher clinical-response scores (Fig. 5h).
Conclusion
Decreases in the cost of sequencing have led to the widespread adoption of integrative sequencing for the study of cancer and precision oncology. Accordingly, our real-time clinical sequencing program was established to explore the practical challenges of clinical translation and at the same time characterize the genomic landscape of advanced cancer. The resulting MET500 cohort represents the first assessment of the genetic and transcriptomic heterogeneity across a wide range of metastatic cancers.
The distribution of mutation frequencies across diverse lineages of metastatic cancers is extremely long-tailed, with relatively few genes mutated at a high rate. We found that 12.2% of our cases harbored potentially pathogenic germline variants, a majority of which (75%) were related to DNA repair pathways. Mutations in DNA repair pathways have therapeutic implications; hypermutated tumors may respond to immune checkpoint inhibitors42, while HR deficiency could suggest sensitivity to PARP inhibitors42,43. The high prevalence of likely pathogenic germline variants suggest that metastatic patients should be considered for genetic counseling and associated germline testing.
By integrating whole exome sequencing with RNA sequencing we were able demonstrate that transcriptome profiling provides clinically important and complementary molecular information. We demonstrate how RNA sequencing can be employed in a clinical context to characterize gene fusions, outlier gene expression, transcriptional pathways and the immune microenvironment. Across the MET500 cohort, 37% cases harbored a putative driver fusion, or an inactivating fusion in a tumor suppressor gene. RNA-seq data played an important role in characterizing the transcriptional networks active in tumor cells as well as the metastatic tumor microenvironment and suggest that metastatic tumors are significantly dedifferentiated, but retain some tissue- and cancer-specific gene expression patterns. We were able to delineate two distinct types of metastases: proliferative and EMT-like. Interestingly, proliferative tumors were associated with increased metabolism and stress response, while EMT-like tumors were associated with inflammation-related signatures.
Particularly valuable in the context of immunotherapy are mechanism-driven biomarkers that delineate discrete immune checkpoints or mechanisms of immune evasion. However, immune biomarkers need to characterize a complex disease state comprising the tumor genotype (e.g., mutational burden), phenotype (e.g., PD-L1 expression), and host response (e.g., presence of CD8+ T-cells). Towards comprehensive immunogenomic profiling, in this study we leveraged DNA and RNA sequencing data, which enabled us to characterize not only the tumor genotype, but also the phenotype of the host-immune response. Our results demonstrate the feasibility of using RNA-seq data to delineate immunologically and potentially clinically distinct subtypes of metastatic tumors, highlighting the potential of clinical RNA sequencing for monitoring the tumor microenvironment and guiding immunotherapeutic approaches.
While this study compares the molecular attributes of a metastatic cancer cohort to those of primary cancer cohorts, it does not utilize matched samples of primary and metastatic biopsies from individual cases. The sequencing of matched samples could illuminate further the processes behind tumor evolution, resistance to therapy, and immune interactions.
In summary, the metastatic solid tumor cohort represented in this study is a powerful complement to studies that have been carried out on primary cancers. Metastatic cancer is a highly heterogeneous disease at the genetic, transcriptomic, and microenvironment level. Significant progress in the treatment of advanced cancer will therefore depend on our ability to learn the therapeutic implications of metastatic heterogeneity and to develop screening methods and clinical trial designs that match patients to the most promising therapies.
METHODS
No statistical methods were used to predetermine sample size. The experiments were not randomized.
Patient enrollment
Sequencing of clinical samples was performed under our Institutional Review Board (IRB)–approved studies at the University of Michigan (Michigan Oncology Sequencing Protocol, MI-ONCOSEQ, IRB # HUM00046018, HUM00067928, HUM00056496). Medically qualified patients 18 years or older with metastatic disease (including advanced or refractory) that could be safely accessed by image-guided biopsy were eligible for inclusion. The MI-ONCOSEQ study was initiated in 2010 and is ongoing as of Feb 2017. All patients provided written informed consent to obtain fresh tumor biopsies, and to perform comprehensive molecular profiling of tumor/germline exomes and tumor transcriptome. 422 patients (MO prefix) were enrolled under the Mi-Oncoseq protocol which included meeting with genetic counselling at the time of consent. A ‘flexible default’ consent model was employed to MO subjects which mandated disclosure of findings that directly impacted the current cancer management, but patients could choose whether to receive incidental results, including those with possible significance for family members or conditions unrelated to the current cancer. The remaining patients (TP prefix) were usually from external sites and were enrolled under a Tissue Profiling protocol without initial genetic counselling. TP subjects were not given the option to decline germline findings, and germline results relevant to cancer were automatically reported. Once sequenced, a patient’s clinical course was updated quarterly in order to document clinical status and treatment decisions made by the physicians since last follow up.
Tissue acquisition and Pathology Review
Needle biopsies or surgically resected tissues were flash frozen in OCT and a section was cut for evaluation. Remaining portions of each specimen were retained for nucleic acid extraction. Hematoxylin and eosin (H&E) stained frozen sections were reviewed by pathologists to identify cores or areas with highest tumor content. In general, multiple sources of data were utilized in confirming the diagnosis and site of origin of the carcinomas, especially metastatic adenocarcinoma in these biopsies sent for NGS. These included review of the electronic medical record for the clinical history, documentation of primary diagnosis and/or previously performed metastatic biopsy with confirmation of site of origin and the morphological assessment of haematoxylin and eosin (H&E)-stained sections (frozen section slides in all cases) and review of concurrent formalin fixed paraffin embedded sections, as the majority of these cases had a prior documentation of metastasis with confirmation of site of origin. In no case was the frozen section slides used exclusively to determine site of origin. Immunohistochemistry using a panel of antibodies was performed on the formalin fixed paraffin embedded sections, especially if the biopsy was the first documentation of metastasis. The antibodies used for confirmation of site of primary origin were based on primary diagnosis; however, when clinically indicated, depending on site of metastasis, presence of multiple primaries and/or if there was a long time gap between primary diagnosis and metastatic biopsy, to the best possible extent other sites of origin were excluded. Presentation at the Precision Medicine Tumor Board included a review of morphological assessment and immunohistochemical results and correlation of these results with expression analysis of the RNA-SEQ data, as well as mutation signatures. Sequencing results were used in a supportive fashion to reinforce the diagnosis of primary origin of the metastatic cancer.
Integrative Clinical Sequencing
Integrative clinical sequencing was performed using standard protocols in our Clinical Laboratory Improvement Amendments (CLIA) compliant sequencing lab10,15. Tissues with highest tumor content for each case were disrupted by 5mm beads on a Tissuelyser II (Qiagen). Tumor genomic DNA and total RNA were purified from the same sample using the AllPrep DNA/RNA/miRNA kit (Qiagen). Matched normal genomic DNA from blood, buccal swab or saliva was isolated using the DNeasy Blood & Tissue Kit (Qiagen). RNA integrity was measured on an Agilent 2100 Bioanalyzer using RNA Nano reagents (Agilent Technologies). RNA sequencing was performed either by poly(A)+ transcriptome or exome-capture transcriptome platform10,44. Both poly(A)+ and capture transcriptome libraries were prepared using 1~2 ug of total RNA. Poly(A)+ RNA was isolated using Sera-Mag oligo(dT) beads (Thermo Scientific) and fragmented with the Ambion Fragmentation Reagents kit (Ambion, Austin, TX). cDNA synthesis, end-repair, A-base addition, and ligation of the Illumina index adapters were performed according to Illumina’s TruSeq RNA protocol (Illumina). Libraries were size-selected on 3% agarose gel. Recovered fragments were enriched by PCR using Phusion DNA polymerase (New England Biolabs) and purified using AMPure XP beads (Beckman Coulter). Capture transcriptomes were prepared as above without the up-front mRNA selection and captured by Agilent SureSelect Human all exon v4 probes following the manufacturer’s protocol. Library quality was measured on an Agilent 2100 Bioanalyzer for product size and concentration. Paired-end libraries were sequenced by the Illumina HiSeq 2000 or HiSeq 2500 (2×100 nucleotide read length), with sequence coverage to 40~50M paired reads. Reads that passed the chastity filter of Illumina BaseCall software were used for subsequent analysis.
Exome libraries of matched pairs of tumor / normal DNAs were prepared as described before10, 15. In brief, 1~3 ug of genomic DNA was sheared using a Covaris S2 to a peak target size of 250 bp. Fragmented DNA was concentrated using AMPure beads, followed by end-repair, A-base addition, ligation of the Illumina indexed adapters, and size selection on 3% Nusieve agarose gels (Lonza). Fragments between 300 to 350 bp were recovered, amplified using Illumina index primers, and purified by AMPure beads. 1 ug of the library was hybridized to the Agilent SureSelect Human All Exon v4. The targeted exon fragments were captured and enriched following the manufacturer’s protocol (Agilent). Paired-end whole exome libraries were analyzed by Agilent 2100 Bioanalyzer and DNA 1000 reagents and sequenced using the Illumina HiSeq 2000 or HiSeq 2500 (Illumina Inc. San Diego, CA).
We used the publicly available software FastQC to assess sequencing quality. For each lane, per-base quality scores across the length of the reads were examined. Lanes were deemed passing if the per-base quality score boxplot indicated that >75% of the reads had >Q20 for bases 1–80. In addition to the raw sequence quality, the alignment quality was also assessed using the Picard package. This allows monitoring of duplication rates and chimeric reads that may result from ligation artifacts - crucial statistics for interpreting the results of copy number and structural variant analysis.
T-cell receptor β repertoire deep sequencing (immunoSEQ): Amplification and sequencing of [TCRB / IGH / IGKL / TCRAD / TCRG] CDR3 was performed using the immunoSEQ Platform (Adaptive Biotechnologies®, Seattle, WA). Same DNA aliquot obtained from frozen tumor tissues was used as for the exome sequencing. The immunoSEQ Platform combines multiplex PCR with high throughput sequencing and a sophisticated bioinformatics pipeline for [TCRB / IGH / IGKL / TCRAD / TCRG] CDR3 analysis that includes internal PCR amplification controls. Duplicate PCR reactions have been done on all samples with >1µg of DNA. Computational analysis of sequencing data, including the estimation of the total number of templates, identification and identification of clonotypes was performed using the vendor-supplied analysis portal.
Mutation analysis
Whole-exome sequencing was performed on Illumina HiSeq 2000 in paired-end mode and the primary base call files were converted into FASTQ sequence files using the bcl2fastq converter tool bcl2fastq-1.8.4 in the CASAVA 1.8 pipeline. The FASTQ sequence files generated were then processed through an in-house pipeline constructed for whole-exome sequence analyses of paired cancer genomes. The sequencing reads were aligned to the reference genome build hg19, GRCh37 using Novoalign Multithreaded (Version 2.08.02) (Novocraft) and converted into BAM files using SAMtools (Version 0.1.18). Sorting and indexing of BAM files utilized Novosort threaded (Version 1.00.01) and duplicates reads were removed using Picard (Version 1.74). Mutation analysis was performed using VarScan2 algorithms (Version 2.3.2) utilizing the pileup files created by SAMtools mpileup for tumor and matched normal samples, simultaneously performing the pairwise comparisons of base call and normalized sequence depth at each position. For single nucleotide variant detection, filtering parameters including coverage; variant read support, variant frequency, P-value, base quality, homopolymer, and strandedness are applied. For indels analysis Pindel (Version 0.2.4) was used on tumor and matched normal samples and indels common in both samples were classified as germline and indels present in tumor but not in normal were classified as somatic. Finally, the list of candidate indels as well as somatic and/or germline mutations was generated by excluding synonymous SNVs. ANNOVAR69 was used to functionally annotate the detected genetic variants and positions are based on Ensemble66 transcript sequences.
Tumor content for each tumor exome library was estimated from the sequence data by fitting a binomial mixture model with two components to the set of most likely SNV candidates on 2-copy genomic regions. The set of candidates used for estimation consisted of coding variants that (1) exhibited at least 3 variant fragments in the tumor sample, (2) exhibited zero variant fragments in the matched benign sample with at least 16 fragments of coverage, (3) were not present in dbSNP, (4) were within a targeted exon or within 100 base pairs of a targeted exon, (5) were not in homopolymer runs of four or more bases, and (6) exhibited no evidence of amplification or deletion. In order to filter out regions of possible amplification or deletion, we used exon coverage ratios to infer copy number changes, as described below. Resulting SNV candidates were not used for estimation of tumor content if the segmented log-ratio exceeded 0.2 in absolute value. Candidates on the Y chromosome were also eliminated because they were unlikely to exist in 2-copy genomic regions. Using this set of candidates, we fit a binomial mixture model with two components using the R package flexmix, version 2.3.8. One component consisted of SNV candidates with very low variant fractions, presumably resulting from recurrent sequencing errors and other artifacts. The other component, consisting of the likely set of true SNVs, was informative of tumor content in the tumor sample. Specifically, under the assumption that most or all of the observed SNV candidates in this component are heterozygous SNVs, we expect the estimated binomial proportion of this component to represent one-half of the proportion of tumor cells in the sample. Thus, the estimated binomial proportion as obtained from the mixture model was doubled to obtain an estimate of tumor content.
Recurrently mutated genes were classified as putative oncogenes and tumor-suppressors. Initially, we divided them into tumor suppressors and oncogenes based on the proportion of inactivating (two-hit, non-sense) aberrations (relative to all other aberration including missense mutations and amplification) using a heuristic cut-off 0.65. This initial classification was then reviewed based on the distribution of somatic mutations in COSMIC (e.g. presence of hotspots, ratio of inactivating mutations, prevalence of frameshifts, etc.), whether the mutations are putative gain or loss-of-function, and relevant gene-related literature. This resulted, for a number of oncogenes but not tumor-suppressors, in the re-classification to tumor suppressor i.e. FAT1, KMT2B, KMT2C, KMT2D, RAD50, RNF43, MSH2, SMC4, KEAP1, MUTYH, BRIP1, and VHL.
Copy number aberrations (CNA)
CNA was quantified and reported for each gene as the segmented normalized log2-transformed exon coverage ratios between each tumor sample and matched normal sample. To account for observed associations between coverage ratios and variation in GC content across the genome, lowess normalization was used to correct per-exon coverage ratios prior to segmentation analysis. Specifically, mean GC percentage was computed for each targeted region, and a lowess curve was fit to the scatterplot of log2-coverage ratios vs. mean GC content across the targeted exome using the lowess function in R (version 2.13.1) with smoothing parameter f=0.05. Partially redundant sequencing of areas of the genome affords the ability for cross validation of findings. We cross-validated exome-based point mutation calls by manually examining the genomic and transcriptomic reads covering the mutation using the UCSC Genome Browser. Likewise, gene fusion calls from the transcriptome data can be further supported by structural variant detection in the genomic sequence data, as well as copy number information derived from the genome and exome sequencing.
Mutation burden estimation
The Varscan2 processed VCF files from 33 TCGA cohorts were downloaded from the GDC data portal and were lifted over from GRCh38 to GRCh37 reference genome using CrossMap to compare with MET500. The mutations were filtered by coverage (at least 10X) and variant allelic fraction (at least 6%). These mutations were further narrowed down to be within 10bp of the Agilent All Exon V4 captured regions. The mutation burden was estimated as (total mutation / total covered bases) *1e6. Finally, we identified 20 common cohorts between MET500 and TCGA.
Comparisons of gene-level aberration frequency
In order to compare the mutation frequency between primary and metastatic tumors we first identified 20 analysis cohorts (tumor types) shared between the TCGA and MET500 projects. For each of those cohorts we obtained aberration frequencies for selected most recurrently/ubiquitously mutated oncogenes (TP53, PTEN, RB1) and tumor suppressors (KRAS, PIK3CA, GNAS). We compare the aberration frequencies for each gene within each tumor type using a Fisher’s exact test for a total of 120 dependent tests. To correct for multiple testing, we applied the BY method of Benjamini, Hochberg, and Yekutieli.
RNA-Seq data analysis
Strand-specific RNA-seq libraries were analyzed using the CRISP clinical RNA-seq pipeline, which comprises: expression analysis, virus detection, and structural variant detection using a separate tool CODAC (manuscript in preparation). CRISP is composed of several tasks: pre-alignment QC, read grooming, alignment, post-align QC, quantification. Notably, fusion-calling is not a part of CRISP and is done independently using multiple of CRISP output files. The components were either chosen based on their performance and robustness (e.g. featureCounts) or rewritten from scratch (HPSEQ, sepath, PaPy). Briefly, reads that pass vendor QC thresholds have been trimmed of adapter sequences and aligned to the GRCh38 reference genome with added sequences for known oncogenic viruses and a transcript reference database based on Gencode V23. STAR_2.4.0g1 was been used for alignment with the following settings not-default: outSAMstrandField:None; alignSJoverhangMin : 8; alignSJDBoverhangMin : 3; scoreGenomicLengthLog2scale : 0; alignIntronMin : 20; alignIntronMax : 1000000; alignMatesGapMax : 1000000. For Chimeric alignment, used for structural variant detection, the following settings were applied: alignIntronMax:400000; alignMatesGapMax:400000; chimSegmentMin:10; chimJunctionOverhangMin:1; chimScoreSeparation:0; chimScoreJunctionNonGTAG:0; chimScoreDropMax:1000; chimScoreMin:1.
The chimeric output was analyzed for chimeric junction supported by spanning and encompassing reads, and then filtered. CRISP and CODAC were tuned to perform optimally with a custom set of reference transcripts based on Gencode V23 (MOTR). This set lacks many questionable transcripts such as non-coding transcripts overlapping coding exons, transcripts linking two protein coding genes, read-through transcripts, non-coding transcripts for protein-coding genes, strange isoforms with extremely long exons, intron-retention isoforms, etc. Many of those can decrease the reliability of gene-based expression estimates and most importantly limit the ability to detect fusions as the detection of chimeric reads rests on the assumption that the “fused” genes are not part by a known isoform. Also by eliminating coding - non-coding overlaps, the coding (PROT) and non-coding (NONC) portions of MOTR are disjoint which simplifies many downstream analyses. In addition to standard chromosomes and unplaced contigs, we included a number of sequences of laboratory contaminants (e.g. Mycoplasma), pathogens (e.g. tuberculosis), and oncogenic viruses (e.g. HPV). A custom pipeline inspects the reads aligned to viral sequences (which are often problematic e.g. highly repetitive). The sequencing approach used in this study does not detect HIV with reliability or sensitivity, precluding its use clinically in this regard. Paired-end reads are trimmed from adapter sequences (in-house tool) and processed twice, once for linear alignment and downstream expression profiling and a second time for chimeric alignment. Before chimeric alignment the reads are “merged”, i.e. if the two mates overlap because the sequenced RNA fragment was shorter than twice the read-length the reads are combined into a synthetic single long read. This greatly improves the sensitivity by which STAR can detect a chimeric junction. CODAC can use both alignment files to call fusions.
Fusion Calling (CODAC)
Our fusion calling pipeline allows us to detect fusions regardless of the location of breakpoints within gene bodies, which in turn allows us detect a wider range of aberrations including gain-of-function fusions and truncating loss-of-function fusions. The chimeric alignments from STAR are aggregated using custom software (in preparation) and filtered for recurrent artifacts, breakpoints within problematic repetitive regions, segmental duplications, and possible alignment errors (mismatches, pseudogenes). Variable cut-offs of supporting reads were required, depending on the breakpoint position (higher if breakpoint is in problematic regions), with a minimum of 3 high-quality spanning reads defined as having a long alignment (>60bp) on both ends of the breakpoint and a low number of mismatches (sequencing errors, SNPs, or mutations), and low repetitiveness score.
Computational Fusion Validation
We carried out both computational and experimental validations to estimate the specificity of our fusion calling algorithm CODAC (manuscript in preparation). First, we ran the algorithm on 50 randomly selected GTEX libraries (normal samples) and found that for 90% of the cases at most 2 false-positive fusions were called. Next, we compared the quality of clinically-reportable or pathogenic fusion calls to the remaining fusion calls, using a compound fusion-quality score which takes into account a number of quality metrics: number of spanning reads, alignment quality, repetitiveness of the DNA, presence of splice donor-acceptor motif etc. Likely pathogenic and non-pathogenic fusions were very similar in terms of overall quality. To further validate our algorithm, we plotted the number of fusions and number of copy-number breakpoints per-sample, and observed that the number of DNA and RNA-breakpoints is highly correlated.
Experimental Fusion Validation
To validate the sensitivity/specificity of the fusion-calling pipeline, we randomly selected 17 private pathogenic fusions on Supplementary Table 6, and 40 fusions from 2 cases with highly rearranged genomes (20 random fusions per case, ranging from high to low supporting reads) and performed RT-PCR followed by Sanger sequencing of the fusion fragments. cDNA was synthesized using the SuperScript III First-Strand Synthesis SuperMix Kit according to the manufacturer’s instructions (Thermo Fisher Scientific/Invitrogen). PCR amplification was performed using fusion-specific primers (synthesized by IDT) and the HotStarTaq Plus Master Mix Kit (QIAGEN). PCR products were subjected to electrophoresis and purified using the QIAEX II Gel Extraction Kit (QIAGEN) before Sanger sequencing. Sanger sequencing was performed by the University of Michigan DNA Sequencing Core. 56 out of the 57 candidates were validated (98.2%) by this approach.
Adjustment
Capture and polyA expression levels can be almost perfectly adjusted across the whole dynamic range of gene expression. The adjustment is based on the (shown valid) assumption that the majority of the differences is due to systematic differences e.g. capture efficiency or transcript stability. These differences were estimated from data of 400 paired polyA and capture libraries using a linear model with shrinkage and variance pooling using limma with Voom precision estimates (manuscript in preparation). Correction of systematic biases through a linear model (Supplementary Methods) eliminated almost all of the apparent differences, and resulted in a good correlation between capture and polyA RNA-seq data (r=0.97) indicating high overall reproducibility, and enabling us to jointly analyze both data sets.
Selection of Marker genes
We followed a multi-step procedure to identify expression markers for normal tissues and primary tumor types. First, we assembled expression compendia for both tasks. The normal tissue compendium included all of the data available from GTEX and the Human Proteome Atlas, for a total of 36 different tissues / organs. The primary cancer compendium included all data from the TCGA for 33 primary tumor types. Then for each tissue / cancer type we sought to find protein-coding genes that fulfill the following criteria:
highly expressed for that given tissue / cancer type
up-regulated in that tissue compared to other tissues / cancer types
expressed in only a few tissues / cancer types
not redundant with other better markers for that tissue / cancer types
We implemented an algorithm (available at: https://github.com/mcieslik-mctp/) that identifies such genes using two statistical criteria: enrichment Z-score (i.e. how much higher a gene is expressed in a target tissue/tumor relative to other tissues tumors) and Hoyer’s sparsity (i.e. sparsity is highest if a gene is expressed only in one tissue type and lowest if it is expressed in all tissues). Next, all genes are ranked according to both measures and an average rank is computed. This ranked list of genes is traversed from the putative markers (highest ranks) to worst (lowest rank) in order to populate a short-list of uncorrelated markers, beginning with the top-ranking gene which is automatically included in the short-list. For each subsequent evaluated marker a correlation to all other previously included markers is computed and a cutoff is applied (Spearman rho>0.85). This process is repeated until 50 short-listed marker genes are identified for each tissue. We have applied this algorithm to the two datasets (i.e. 36 normal tissues and 33 cancer types) which resulted in two sets of markers genes Nt36 and Tc33, respectively.
Expression Signature Analysis
To estimate signature/hallmark/pathway activity levels we used two different approaches. The first relative (“intrinsic”) approach is similar to GSVA and ssGSEA in that it estimates the activity of a pathway in one sample relative to the activity of that pathway in a cohort of other samples. In our case the cohort is the MET500. The expression of each gene in the pathway was transformed into percentiles and the activity of each pathway was calculated as the average percentile score of all genes in a pathway - 50 (i.e. the expected median activity of a pathway). The intrinsic score has been shown to correlate very well with GSVA estimates. All GSVA analyses have been carried out using default settings. The second “extrinsic” approach is analogous to the relative (intrinsic) approach, but expression percentiles for each gene are calculated, not relative within the MET500 cohort, but compared to the expression levels of that gene in over 8000 samples from 36 normal tissues (GTEX and HUPA), each tissue represented by the same number of transcriptomes (oversampling for some of the less well studied tissues). For all pathway analyses we used the gene sets provided by MSigDB, including the Hallmark sets, and the experimentally perturbed data sets.
MImmScore
The MImmScore is an aggregate measure of immune infiltration based on the expression of multiple immune-related genes. It is derived from the “immune signature” genes used in the ESTIMATE method by (Yoshihara et. al, 2013). The 141 genes included in this set fulfill a number of criteria: (i) are common to RNA-seq and major microarray platforms, (ii) are highly expressed in hematopoietic cells compared to normal tissues (iii) are up-regulated in highly immune infiltrated ovarian tumors, (iv) are non-redundant. A signature derived from the expression of these genes has been shown to track with the amount of non-tumor cells based on EpCam expression. We have used all of the 141 “immune score” genes, but chose a different statistical approach to transform their relative expression levels into a compound score. We used the so called “inverse normal transformation” method common in eQTL and other regression analyses. Briefly, this rank-based method minimizes the influence of outliers, and genes with non-normally distributed expression levels, on the compound score. The first step is to transform the variable (i.e. gene expression across samples) to a ranks and subsequently percentiles. The percentiles then transformed to standard normal deviates using the inverse normal (or probit function). In other words, the expression of each gene is made to follow the normal distribution. The MImmScore is simply computed by summing, for each sample, the standard normal deviates for each gene. The MImmScore is hence analogous to the “intrinsic” signature expression analysis, with the added step of converting percentiles to the Z-scores associated with a given probability (using the standard qnorm function in R). Samples with high levels of immune infiltration will have many genes expressed above the 50th percentile (Z-score > 0) and hence the overall MImmScore will be positive.
Data and Code availability
To make the data accessible to the broader scientific and clinical communities, we have made the somatic landscape searchable through a MAGI-based web interface45 at: http://met500.path.med.umich.edu.
All custom analysis software used in this study are publically available on github:
Pathogenic germline variant analysis
Pathogenicity of germline variants were determined through review of the published literature, public databases including but not limited to ClinVar, Human Genome Mutation Database, and Leiden Open Variation Databases, and variant specific databases (e.g., International Agency for Research on Cancer TP53 Database, International Society for Gastrointestinal Hereditary Tumors mutation databases). Only cancer relevant germline variants that had been previously categorized as pathogenic in ClinVar, or adjudicated at the Precision Medicine Tumor Board as pathogenic, were disclosed on the clinical report. These clinically reported germline variants are shown in Supplementary Table 4. Variants with conflicting pathogenicity reports and variants not previously reported were considered to be of uncertain significance and were not considered for disclosure. Following disclosure, familial testing was recommended. Clinical relevance of somatic variants was investigated using an integrated approach incorporating technical considerations, (recurrence, variant allele fraction, expression levels, and predictive algorithms for pathogenicity), variant specific information (ClinVar, published literature, and curated gene specific resources), as well as published correlations of drug / variant sensitivity profiles. Considerations of tumor heterogeneity, including clonal versus subclonal mutations were addressed by comparing variant allele fractions and copy number estimates for each of the mutations to post-sequencing estimates of tumor content derived from SNV and copy number analyses.
Precision Medicine Tumor Board (PMTB) activity
A bi-weekly, multi-disciplinary PMTB interpreted and deliberated on sequencing results for each patient. PMTB participants included pediatric and adult oncologists, geneticists, pathologists, biologists, bioinformaticians, bioethicists, genetic counselors, study coordinators, and ad hoc expertise. Selected findings underwent additional independent CLIA-validated testing, and summarized results were disclosed to treating oncologists and families by the clinical sequencing team, board certified clinical geneticists, and/or counselors, as appropriate. For the purposes of this study potentially actionable findings (PAF) were defined as any genomic findings discovered during sequencing analysis that could lead to a 1) change in patient management by providing a targetable molecular aberration, 2) change in diagnosis or risk stratification or 3) provides cancer-related germline findings which inform patients/families about a potential future risk of various cancers.
Extended Data
Extended Data Figure 1. Demographics of the MET500 cohort and summary of common genetic aberrations.
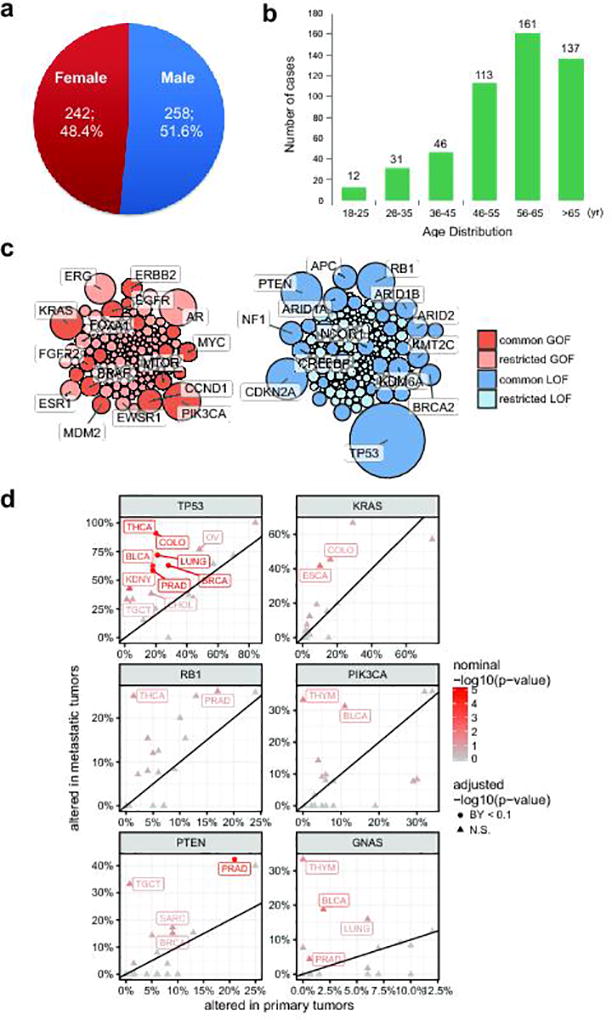
a. Gender distribution of the MET500 cohort. b. Age distribution of the MET500 cohort. c. Bubble plot of clinically actionable genetic aberrations. Genes have been divided by putative gain-of-function (oncogene, red) or loss-of-function (tumor suppressor, blue) status. Common aberrations are defined as those observed in 5 or more MET500 analysis cohorts (Fig 1c), restricted aberrations are found in less than five analysis cohorts. Bubble area is proportional to the observed frequency of the aberration across the MET500 cohort. d. Comparison of genetic aberration frequencies (SNVs, indels, amplifications, predicted homozygous deletions) between primary (TCGA) and metastatic (MET500) tumors for select tumor-suppressors (left panels) and oncogenes (right panels). TCGA data for the primary cancer cohorts have been obtained from the cBio portal. Nominal statistical significance is based on the Fisher’s exact test. Statistically significant differences in frequencies following correction for multiple dependent tests using the Benjamini-Yekutieli procedure are indicated as circles, insignificant differences are shown as triangles.
Extended Data Figure 2. Analysis of pan-cancer metastatic transcriptomes.
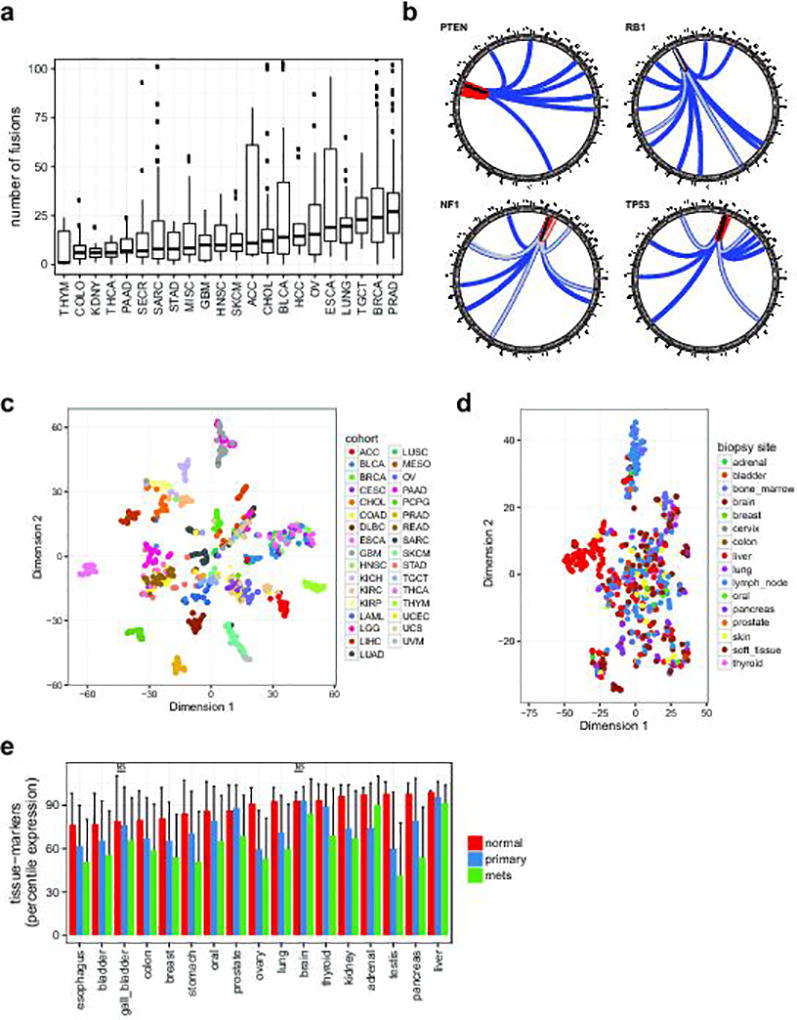
a. Structural rearrangements in metastatic genomes. Distribution of the number of fusions per case is plotted across the MET500 by analysis cohort (see Fig. 1c for cancer abbreviations). Y-axis is truncated at 100 fusions. b. Summary circos diagrams of predicted inactivating fusions for select tumor suppressor genes across the cohort. Arc end positions indicate the chimeric junctions; colors indicate type of rearrangement. Black: tandem duplication, blue: translocation, red: inversion, gray: signifies that multiple close junctions were detected. c. t-SNE plot for the TCGA pan-cancer meta-cohort (a random selection of cases from each primary tumor type) based on the expression of tumor-type specific marker genes (same genes as in Fig. 4a). d. t-SNE plot for the MET500 samples colored by biopsy site (same samples as in Fig. 4a, there colored by cancer type). e. Average percentile expression of tissue-specific genes in normal tissues, primary cancers, and metastases. Error-bars indicate standard deviation. Significance test have been carried out for all normal-primary and primary-mets pairs of samples, all comparisons were significant (p<0.01) according to a two-tailed t-test, with the exception of those indicated with (NS). Abbreviations: ACC, Adrenocortical Carcinoma; BLCA, Bladder Urothelial Carcinoma; BRCA, Breast Invasive Carcinoma; CESC, Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma; CHOL, Cholangiocarcinoma; COAD, Colon Adenocarcinoma; DLBC, Lymphoid Neoplasm Diffuse Large B-cell Lymphoma; ESCA, Esophageal Carcinoma; GBM, Glioblastoma Multiforme; HNSC, Head and Neck Squamous Cell Carcinoma; KICH, Kidney Chromophobe; KIRC, Kidney Renal Clear Cell Carcinoma; KIRP, Kidney Renal Papillary Cell Carcinoma; LAML, Acute Myeloid Leukemia; LGG, Brain Lower Grade Glioma; LIHC, Liver Hepatocellular Carcinoma; LUAD, Lung Adenocarcinoma; LUSC, Lung Squamous Cell Carcinoma; MESO, Mesothelioma; OV, Ovarian Serous Cystadenocarcinoma; PAAD, Pancreatic Adenocarcinoma; PCPG, Pheochromocytoma and Paraganglioma; PRAD, Prostate Adenocarcinoma; READ, Rectal Adenocarcinoma; SARC, Sarcoma; SKCM, Skin Cutaneous Melanoma; STAD, Stomach Adenocarcinoma; TGCT, Testicular Germ Cell Tumors; THYM, Thymoma; THCA, Thyroid Carcinoma; UCS, Uterine Carcinosarcoma; UCEC, Uterine Corpus Endometrial Carcinoma; UVM, Uveal Melanoma.
Extended Data Figure 3. Global activity of oncogenic signatures.
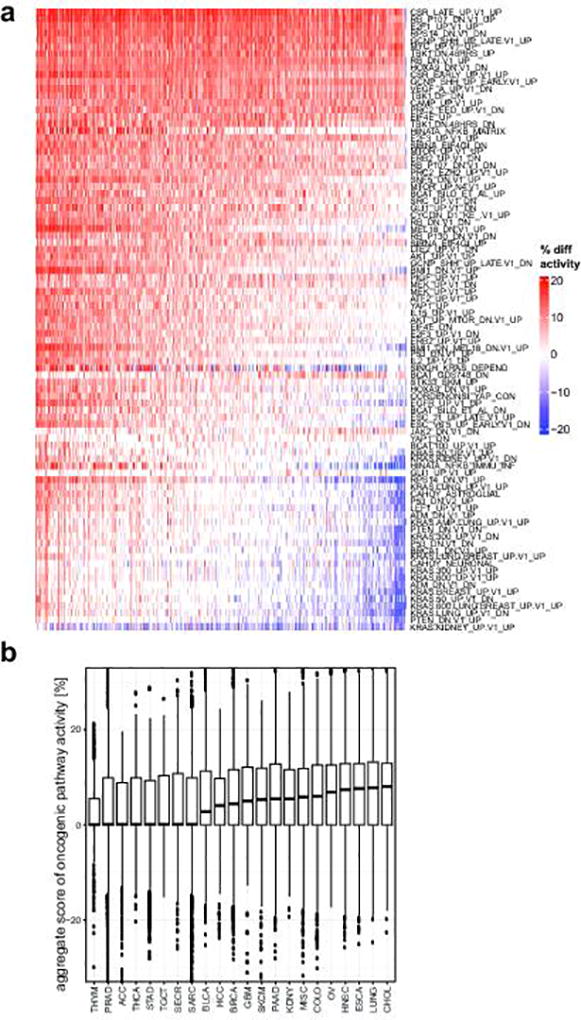
a. Activity of signatures is calculated relative to a normal tissue baseline, i.e. activity scores are compared to a compendium of 36 normal tissues. Therefore, this plot represents a comparison of pathway activities between metastatic tissues and normal tissues. Increased activity (positive difference, red) or decreased activity (negative difference, blue) indicates that the signature genes are on average more (or less) expressed in a metastatic tumor sample relative to the baseline (in average percentile point difference labeled “% diff activity”). Samples (columns) are ordered from left to right by decreasing average activity difference (column averages, i.e. the aggregate score in panel b) b. Boxplots summarizing the aggregate scores (column averages of “% diff activity”) in a. Analysis cohorts are ordered left-to-right by median aggregate scores.
Extended Data Figure 4. Relative activity of oncogenic signatures.
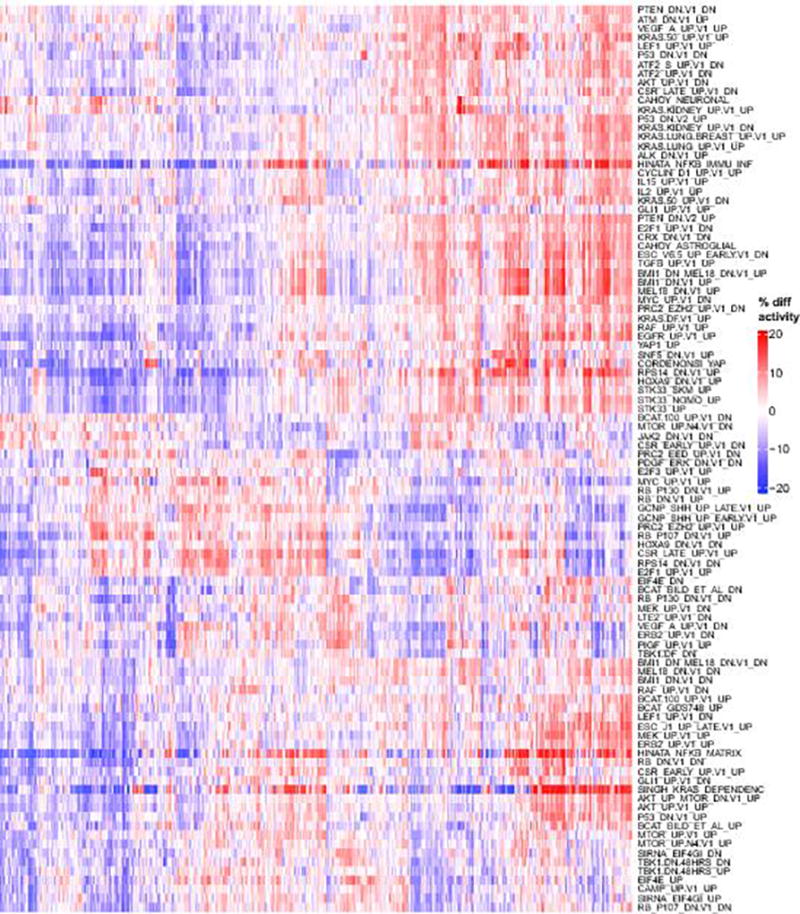
Hierarchically clustered heatmap of activity scores for the most variable oncogenic signatures. In contrast to (Supp. Fig. 7), here activity scores are computed intrinsically, i.e. relative to other samples in the MET500 (like ssGSEA or GSVA), which represents a relative comparison between different patients / samples. Red indicates that a signature is more active (in percentile points) for a given sample relative to the median activity, blue indicates that a signature is less active for a given sample.
Extended Data Figure 5. Activity of cancer hallmarks in metastatic cancers.
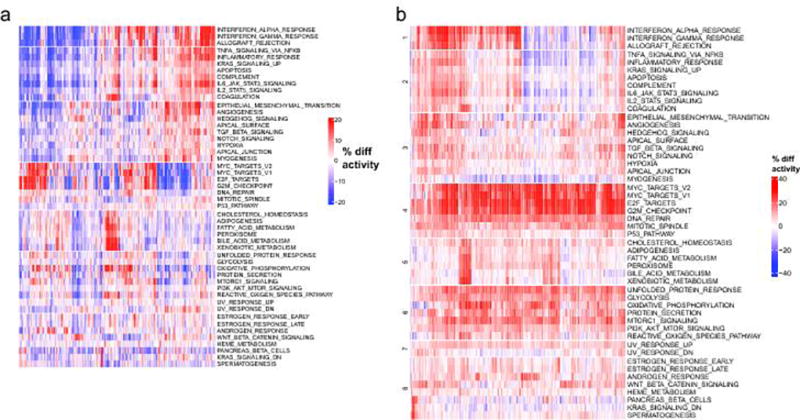
Clustered heatmaps of activity scores for the 50 MSigDB cancer hallmarks are shown. a. Gene expression patterns of cancer hallmark pathways. Average increase (red) or decrease (blue) in the relative expression levels (percentiles) of transcriptional signatures associated with the hallmarks of cancers are illustrated. b. Activity scores are calculated relative to a compendium of 36 normal tissues, which represent a comparison of hallmark activities between metastatic tissues and normal tissues (analogous to Supp. Fig. 7 but for a different gene sets). Red indicates that a signature is more active (in percentile points) for a given sample relative to the median activity, blue indicates that a signature is less active for a given sample.
Extended Data Figure 6. Discovery of oncogenic meta-signatures.
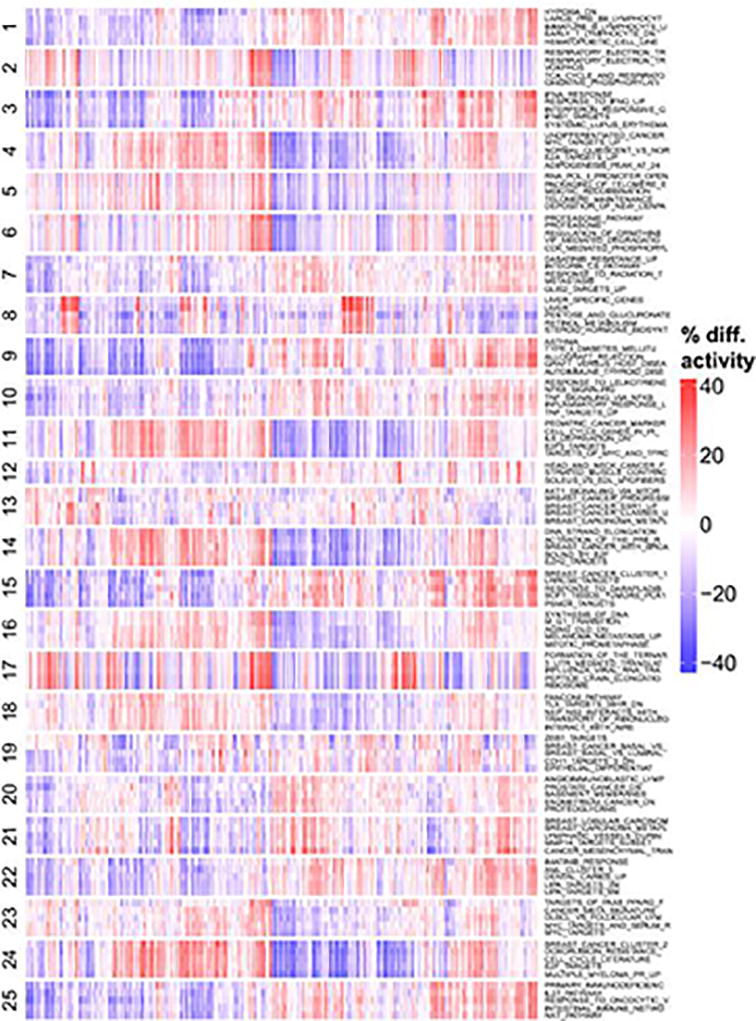
Relative activity scores were computed for all experimental signatures in the MSigDB database across the MET500 cohort. The signatures were clustered into 25 meta-signatures based on their activity profiles across the MET500. For each of the 25 meta-signature clusters, the 5 most variable signatures are selected. Red indicates that a signature is more active (in percentile points) for a given sample relative to the median activity across the MET500. Blue indicates that a signature is less active for a given sample.
Extended Data Figure 7. Activity of the oncogenic meta-signatures.

a. Relative activity of EMT and proliferation signatures across the TCGA analysis meta-cohort. b. Relative activity of the 25 meta-signatures across MET500 samples from different biopsy sites. Red indicates that a signature is more active for a given biopsy site relative to the median activity, blue indicates that a signature is less active for a given biopsy site. c. Relative activity of the 25 meta-signatures across samples from different normal tissues. Red indicates that a signature is more active (in percentile points) for a given tissue relative to the median activity, blue indicates that a signature is less active for a given tissue. d. Correlations between the 25 meta-signatures. Correlation heatmap and hierarchical clustering showed similarities (red) and dissimilarities (blue) in the transcriptional activity of computationally derived aggregate sets of MSigDB signatures, i.e. “meta-signatures” across samples from the MET500 stratified by the most common primary tumor type (left panels) and biopsy site (right panels).
Extended Data Figure 8. Prediction of immune infiltration in cancer tissues.
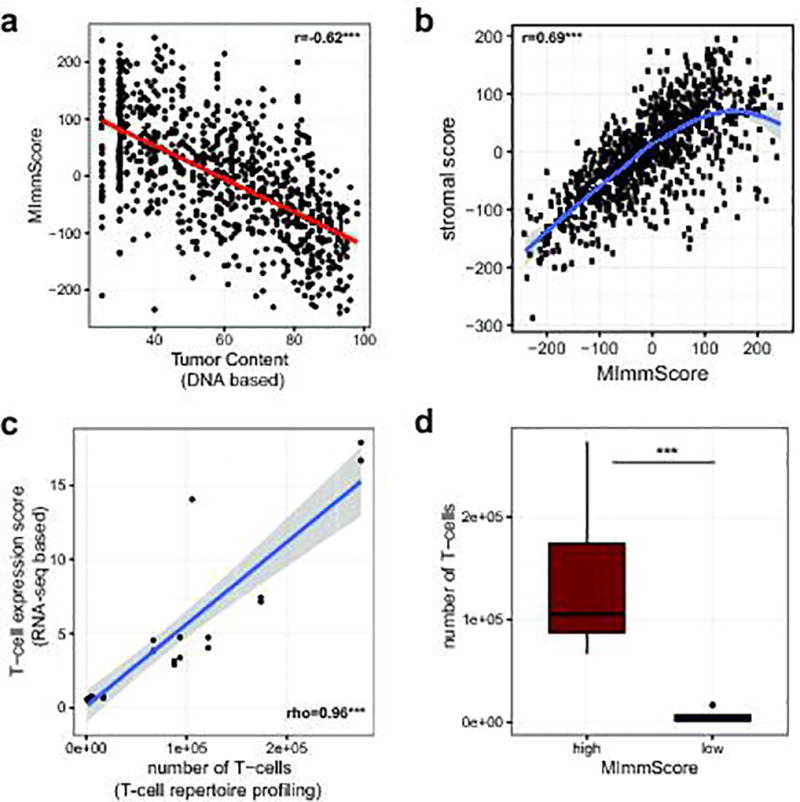
a. Correlation between the MImmScore, a measure of absolute immune infiltration in a tumor samples, with tumor content estimated from exome DNA sequencing using CNVs and SNVs. b. Correlation between MImmScores and an analogous score for tumor-stromal infiltration. c. Correlation between a T-cell expression score summarizing the expression levels (RNA-seq based) of marker genes: CD3D, CD3E, CD3G, CD6, SH2D1A, TRAT1 and the estimated number of T-cells based on T-cell repertoire profiling (DNA-based). d. Number of T-cells based on T-cell repertoire profiling for index cases stratified into MImmScore low (<0) or MImmScore high (>0). Significance levels of Spearman rank correlation coefficient test: * (p=0.05–0.001), ** (p=0.001-1e-6), *** (p<1e-6) test and Wilcoxon test.
Extended Data Figure 9. Differential immune infiltration in various cancer types.
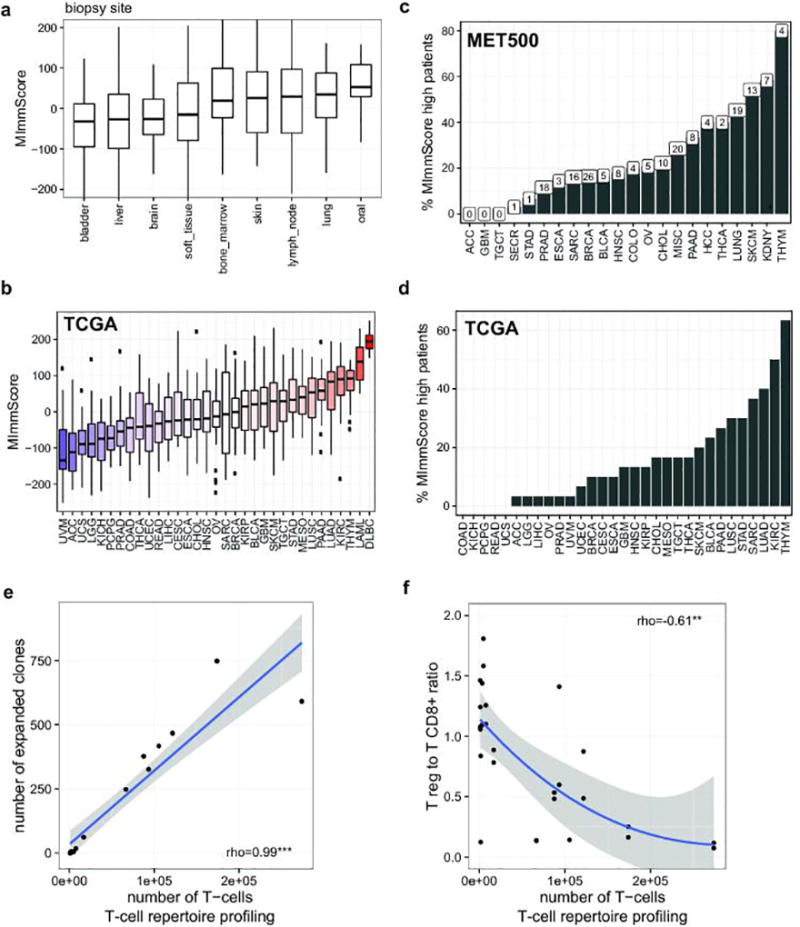
a. Distribution of MImmScores, a measure of the magnitude of immune infiltration in a tumor sample, for MET500 samples/patients grouped by tumor biopsy site. b. Distribution of MImmScores across the TCGA meta-cohort, grouped by primary cancer designation. Hematological malignancies (DLBC, LAML) are included as positive control. c. Percentage of patients in each of the MET500 analysis cohorts that has a high MImmScore defined here as >80th percentile across the whole MET500. The total number of cases with high MImmScore is indicated above each bar. d. Same as c but for the TCGA meta-cohort. e. Correlation between the total number of T-cells (templates) based on T-cell repertoire (DNA) sequencing of the T-cell receptor CDR3 sequence, and the number of expanded clones, an expanded T-cell clone is defined as having more than 30 cells with the same CDR3 sequence. f. Ratio of expression levels for markers of CD8+ T-cells (CD8A, CD8B) and T regs (FOXP3) as a function of the total number of T-cells. Significance levels of Spearman rank correlation coefficient: * (p=0.05–0.001), ** (p=0.001-1e-6), *** (p<1e-6).
Extended Data Figure 10. Genomic correlates of immune infiltration.
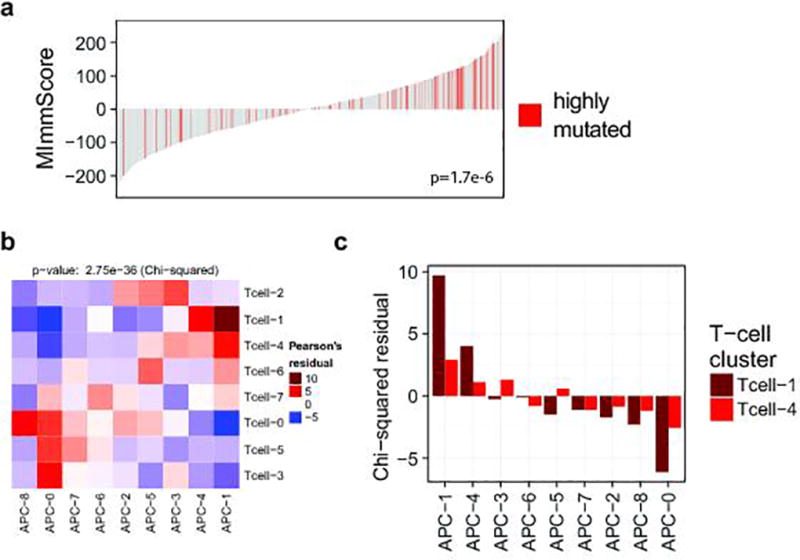
a. Association between the MImmScore and mutation status (hypermutated samples have been defined here as having >250 non-synonymous mutations). Statistical significance of this association was done using logistic regression. b–c. The chi-square test for independence is used to determine whether the clusterings of samples based on T-cell and APC markers are independent. Enrichment or depletion is calculated as the Pearson residual. Red indicates (positive enrichment) that the clusters overlap significantly. Blue indicates (depletion) that clusters tend to be mutually exclusive. Clustered heatmap of enrichment levels (chi-square table cell residuals) is shown in b. Enrichment levels for clusters for the active Tcell-1 and Tcell-4 clusters and all APC clusters (APC-1,4 active) are shown in c.
Supplementary Material
Acknowledgments
Support: This work was supported by a NIH Clinical Sequencing Exploratory Research (CSER) Award NIH 1UM1HG006508. Other sources of support included the Prostate Cancer Foundation, Stand Up 2 Cancer (SU2C)-Prostate Cancer Foundation Prostate Dream Team Grant SU2C-AACR-DT0712, the Early Detection Research Network grant U01 CA214170, and a Prostate SPORE grant P50 CA186786. A.M.C. is a Howard Hughes Medical Institute Investigator, A. Alfred Taubman Scholar, and American Cancer Society Professor. M.C. is supported by a PCF Young Investigator Award.
Additional Contributions: We acknowledge Yu Ning, Rui Wang, Xiaoxuan Dang, Mandy Davis, Lynda Hodges, Janice Griggs, Jyoti Athanikar, Christine Brennan, Christine Betts, Jin Chen, Shanker Kalyana-Sundaram, Karen Giles, and Rohit Mehra, for their contributions to this study. Over 100 physicians referred patients to this study and we would like to acknowledge the following physicians, Kathleen Cooney, Maha Hussain, Susan Urba, Norah Henry, Vaibhav Sahai, Diane Simeone, Chris Lao, Jeffrey Smerage, Megan Caram, Monika Burness, Greg Kalemkerian, Catherine Van Poznak, Max Wicha, Ron Buckanovich, Jose Bufill, Petros Grivas, Patrick Hu, Aki Morikawa, Phil Palmbos, Bruce Redman, Felix Feng, Gary Hammer, Sophia Merajver, Alex Pearson. We thank Sameek Roychowdhury and Ken Pienta for help in protocol development for the Mi-Oncoseq program. Most importantly, the authors would like to recognize the enormous generosity and kindness of the cancer patients and families for participating in this study.
Footnotes
Author Contributions: D.R.R., Y.M.W., and X.C. coordinated clinical sequencing. R.J.L., M.C., P.V. developed the bioinformatics analysis. J.S. coordinated sample procurement, L.P.K., D.L. and S.A.T. led the histopathology analysis. D.C.S., S.S., M.M.Z., A.A., R.C., F.W., L.H.B., R.J.M., N.R., A.F.S. and D.F.H., coordinated patient recruitment. E.R. was the lead study coordinator. J.E., V.M.R., E.M.S. and J.I. provided genetic counseling and assessment of PPGMs and V.J. and K.O. analyzed relative risk assessment. J.S.R. coordinated the bioethics component. M.T. and A.M.C. coordinated IRB protocol development. D.R.R., Y.M.W. and C.K. prepared PMTBs. E.C., M.T., D.F.H., D.R.R. and Y.M.W. implemented the clinical tiering of molecular aberrations. Y.M.W., D.R.R., M.C. and A.M.C. developed the figures and tables. A.M.C., M.C., D.R.R. and Y.M.W. wrote the manuscript with input from all authors. A.M.C. and M.T. designed and supervised the study.
Competing Financial Interests: The authors have no competing financial interests to disclose.
Data deposition: Sequencing data from the 500 patients enrolled in this study can be obtained from dbGAP accession phs000673.v2.p1. The MET500 web portal is available at: http://met500.path.med.umich.edu
References
- 1.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nature reviews. Cancer. 2006;6:449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 2.Steeg PS. Targeting metastasis. Nature reviews. Cancer. 2016;16:201–218. doi: 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman AA, Letai A, Fisher DE, Flaherty KT. Precision medicine for cancer with next-generation functional diagnostics. Nature reviews. Cancer. 2015;15:747–756. doi: 10.1038/nrc4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mauer CB, Pirzadeh-Miller SM, Robinson LD, Euhus DM. The integration of next-generation sequencing panels in the clinical cancer genetics practice: an institutional experience. Genetics in medicine : official journal of the American College of Medical Genetics. 2014;16:407–412. doi: 10.1038/gim.2013.160. [DOI] [PubMed] [Google Scholar]
- 5.Shen T, Pajaro-Van de Stadt SH, Yeat NC, Lin JC. Clinical applications of next generation sequencing in cancer: from panels, to exomes, to genomes. Frontiers in genetics. 2015;6:215. doi: 10.3389/fgene.2015.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones S, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Science translational medicine. 2015;7:283ra253. doi: 10.1126/scitranslmed.aaa7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roychowdhury S, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Science translational medicine. 2011;3:111ra121. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byron SA, Van Keuren-Jensen KR, Engelthaler DM, Carpten JD, Craig DW. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nature reviews. Genetics. 2016;17:257–271. doi: 10.1038/nrg.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352:169–175. doi: 10.1126/science.aaf2784. [DOI] [PubMed] [Google Scholar]
- 10.Robinson D, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research, N et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nature genetics. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gagan J, Van Allen EM. Next-generation sequencing to guide cancer therapy. Genome medicine. 2015;7:80. doi: 10.1186/s13073-015-0203-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mardis ER. The translation of cancer genomics: time for a revolution in clinical cancer care. Genome medicine. 2014;6:22. doi: 10.1186/gm539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsons DW, et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA oncology. 2016 doi: 10.1001/jamaoncol.2015.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mody RJ, et al. Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. Jama. 2015;314:913–925. doi: 10.1001/jama.2015.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagle N, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer discovery. 2012;2:82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pritchard CC, et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. The New England journal of medicine. 2016;375:443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palanisamy N, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nature medicine. 2010;16:793–798. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson DR, et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nature medicine. 2011;17:1646–1651. doi: 10.1038/nm.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nature communications. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agaram NP, Zhang L, Sung YS, Singer S, Antonescu CR. Extraskeletal myxoid chondrosarcoma with non-EWSR1-NR4A3 variant fusions correlate with rhabdoid phenotype and high-grade morphology. Human pathology. 2014;45:1084–1091. doi: 10.1016/j.humpath.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinreb I, et al. Novel PRKD gene rearrangements and variant fusions in cribriform adenocarcinoma of salivary gland origin. Genes, chromosomes & cancer. 2014;53:845–856. doi: 10.1002/gcc.22195. [DOI] [PubMed] [Google Scholar]
- 23.Amir E-aD, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nature Biotechnology. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lonsdale J, et al. The Genotype-Tissue Expression (GTEx) project. Nature genetics. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liberzon A, et al. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Systems. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liberzon A, et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaske CJ, et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;26:i237–i245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.López-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Molecular Medicine. 2009;1:303–314. doi: 10.1002/emmm.200900043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshihara K, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nature communications. 2013;4 doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Attig S, et al. Simultaneous infiltration of polyfunctional effector and suppressor T cells into renal cell carcinomas. Cancer Res. 2009;69:8412–8419. doi: 10.1158/0008-5472.CAN-09-0852. [DOI] [PubMed] [Google Scholar]
- 32.Nakano O, et al. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001;61:5132–5136. [PubMed] [Google Scholar]
- 33.Ruffini E, et al. Clinical significance of tumor-infiltrating lymphocytes in lung neoplasms. Ann Thorac Surg. 2009;87:365–371. doi: 10.1016/j.athoracsur.2008.10.067. discussion 371-362. [DOI] [PubMed] [Google Scholar]
- 34.Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 35.Newman AM, et al. Robust enumeration of cell subsets from tissue expression profiles. Nature Methods. 2015 doi: 10.1038/nmeth.3337. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 37.Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther. 2015;14:847–856. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 38.van Houdt IS, et al. Favorable outcome in clinically stage II melanoma patients is associated with the presence of activated tumor infiltrating T-lymphocytes and preserved MHC class I antigen expression. Int J Cancer. 2008;123:609–615. doi: 10.1002/ijc.23543. [DOI] [PubMed] [Google Scholar]
- 39.Galon J, et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 40.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ulloa-Montoya F, et al. Predictive Gene Signature in MAGE-A3 Antigen-Specific Cancer Immunotherapy. Journal of Clinical Oncology. 2013;31:2388–2395. doi: 10.1200/JCO.2012.44.3762. [DOI] [PubMed] [Google Scholar]
- 42.Mateo J, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. The New England journal of medicine. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nature reviews. Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 44.Cieslik M, et al. The use of exome capture RNA-seq for highly degraded RNA with application to clinical cancer sequencing. Genome research. 2015;25:1372–1381. doi: 10.1101/gr.189621.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leiserson MD, et al. MAGI: visualization and collaborative annotation of genomic aberrations. Nat Methods. 2015;12:483–484. doi: 10.1038/nmeth.3412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
To make the data accessible to the broader scientific and clinical communities, we have made the somatic landscape searchable through a MAGI-based web interface45 at: http://met500.path.med.umich.edu.
All custom analysis software used in this study are publically available on github: