

Abstract
Structural changes in the gut microbial community have been shown to accompany the progressive development of colorectal cancer. In this review we discuss recent hypotheses on the mechanisms involved in the bacteria-mediated carcinogenesis, as well as the triggering factors favoring the shift of the gut microbiota from a mutualistic to a pro-carcinogenic configuration. The possible role of inflammation, bacterial toxins and toxic microbiota metabolites in colorectal cancer onset is specifically discussed. On the other hand, the strategic role of inflammation as the keystone factor in driving microbiota to become carcinogenic is suggested. As a common outcome of different environmental and endogenous triggers, such as diet, aging, pathogen infection or genetic predisposition, inflammation can compromise the microbiota-host mutualism, forcing the increase of pathobionts at the expense of health-promoting groups, and allowing the microbiota to acquire an overall pro-inflammatory configuration. Consolidating inflammation in the gut, and favoring the bloom of toxigenic bacterial drivers, these changes in the gut microbial ecosystem have been suggested as pivotal in promoting carcinogenesis. In this context, it will become of primary importance to implement dietary or probiotics-based interventions aimed at preserving the microbiota-host mutualism along aging, counteracting deviations that favor a pro-carcinogenic microbiota asset.
Keywords: Colorectal cancer, Inflammation, Gut microbiome, Co-abundance groups
Core tip: By performing the co-abundance groups analysis of the publicly available datasets from microbiome surveys in colorectal cancer (CRC) patients, we have been successful in identifying pro-carcinogenic and protective groups of microorganisms, showing the potential to modulate the fate of CRC onset and progression. Possible mechanisms involved in microbiota-dependent carcinogenesis are reviewed, and the central role of inflammation as a trigger forcing the microbiota from a mutualistic configuration to a CRC-promoting asset is discussed. Finally, possible intervention strategies for modulating microbiome in order to preserve its mutualistic configuration along life span are suggested.
HUMAN INTESTINAL MICROBIOTA
Structure of the human intestinal microbiota
Outnumbering human cells 10 to 1, over 100 trillion microbes are hosted in the human body, with the majority of them residing in the gut, in a continuum of dynamic ecological communities, referred to as microbiome[1]. From 101 to 103 microbes per gram of content in the stomach and duodenum, the human gut microbiota reaches a microbial density of 104 to 107 cells per gram in the jejunum and ileum, culminating with 1013-1014 cells in the colon and feces[2,3].
Metagenomic surveys of the intestinal microbiota revealed an immense phylogenetic diversity, estimating more than 1000 species-level phylotypes across the human population, with at least 160 prevalent species per individual[4]. While phylogenetic diversity is high at the species level, most of the endogenous bacteria in healthy adults belong to just two phyla, Firmicutes and Bacteroidetes, which account for > 90% of the known phylogenetic categories of the human gut. Members of Actinobacteria, Proteobacteria, Fusobacteria, Verrucomicrobia, Spirochaetes and Lentisphaerae are regularly present but scarce (< 1%-15%)[5-9].
Since the first application of culture-independent methods a large inter-individual variability in microbial compositions was apparent[4], with twins sharing less than 50% of their species-level microbial taxa[10]. The multiple genetic and environmental factors that contribute to shape the individuality of the gut microbiota composition are now beginning to be understood, reflecting interpersonal, geographical, lifestyle and temporal differences[11-13], and not least, perturbations caused by disease. Recent work has established that despite the unique fingerprint of microbial taxa per individual, a core of > 50 taxa can be found in nearly half of the human subjects sampled[4,8]. It has been suggested that individuals can be categorized into one of three predominant variants or ‘‘enterotypes’’ based on the abundance of dominant genera (Bacteroides, Prevotella or Ruminococcus)[14], though some researchers are now favoring the concept of a continuum or gradient of species functionality rather than a discontinuous variation with segregated types[15]. Individuals have also been shown to share a set of microbial genes involved in central metabolic pathways, and deviations from this functional core have been associated with altered physiological states[16]. However, the subject-specific genetic diversity is remarkable and still remains largely unassigned, with a probably unique metagenomic genotype per individual[17].
Human gut microbiome and role in host physiology
The collective genome of the human intestinal microbiota-microbiome-contains 3.3 million microbial genes, 150-fold more than the human genome[4]. Adding this immense gene catalogue to host genetics, intestinal microorganisms are expected to exert a profound influence on human physiology and metabolism. In fact, gut microbes complement several gaps in our metabolic pathways, e.g., producing essential vitamins and oligo-elements, as well as affording the extraction of energy from otherwise indigestible carbohydrates[18], playing a major role in host energy balance and nutrition[19]. This function has probably been the initial evolutionary force toward the microbiota establishment as an animal and human symbiotic partner[20]. Other recognized functions include the support for colonization resistance against incoming enteropathogens. Mechanism involved in this barrier effect are: competition for food resources[21], inhibition of pathogen growth by means of acetate production[22], killing with bacteriocins[23], and immune response stimulation[24,25]. The gut microbiota also acts as an integral component of the human immune system, finely calibrating the immunological potential and responses at different host ages[26,27]. The intimate interplay between gut microbes and the mucosal immune system has indeed proved to be crucial for immune education during our infancy as well as for maintaining a well-balanced immune homeostasis along the adult life[26,28]. Of note, accumulating data are also supporting the emerging concept of a microbiota-gut-brain axis with a role in the regulation of anxiety, cognition, pain and behavior, and a possible contribution to the pathophysiology of central nervous system disorders[29-32].
Microbiota dynamics in response to diet-inflammation-age
The intestinal microbiota composition was believed to be stable throughout adulthood until few years ago[33,34]. More recently, with the bloom of longitudinal studies in humans, the plasticity of this ecosystem has become evident, highlighting that diet, environment, and physiological changes can impact on both composition and functionality of the gut microbiota[12,27]. Faith et al[35] investigated the normal long-term plasticity of the human gut microbiota in healthy subjects. By applying a low-error amplicon sequencing approach, the Authors demonstrated that 40% of the individual microbiota was variable over the time course of 5 years.
The effect of changes in dietary habits is the plainest manifestation of the ability of the microbiota to adapt its architecture in response to environmental stimuli, with the speed and efficacy required for the maintenance of the nutritional function of the host-microbiota symbiosis. Indeed, short-term dietary responses of the microbiota composition were detected after 24 h and seemed to be driven principally by the type of ingested fermentable carbohydrates[36,37]. These fluctuations could be considered a necessary feature of an intestinal microbial ecosystem able to rapidly adapt itself to the host requirements, maximizing the efficiency of nutrient extraction and supporting health. Remarkably, the same changes in diet in different persons did not result in the same final microbiota configuration since the diet-related variations did not overcome the inter-individual differences. Conversely, in the long term, people with similar dietary patterns may end up sharing a similar architecture of the gut microbiota; in fact, it has been shown that the presence or absence of several bacterial taxa can be associated with the intake of different nutrients[37].
Along with the dietary influence, a certain plasticity of the human intestinal ecosystem is being observed in response to less obvious environmental stressors, such as climate and geography[38,39], as well as the degree of exposure to environmental bacteria, the latter being of primary importance for the education and the maintenance of the functionality of the immune system from birth to adulthood[40-42]. Moreover, the consumption of drugs, especially antibiotics but also anti-inflammatory medicines, impacts on the gut microbiota composition[43-45] and different configurations of the microbiota, in turn, have the ability to promote or reduce the metabolization and effectiveness of drugs[46]. Along adulthood and later in life, natural physiological changes add themselves to the list of drivers of modification in the microbiota structure, both temporarily (i.e., pregnancy or lactation[47]) and permanently, as in the aging process.
Aging can impact on the gut microbiota structure directly, by means of age-related physiological processes involving local and systemic inflammation (i.e., immunosenescence and inflamm-aging; see below), and indirectly, causing changes in dietary habits and lifestyle[48]. Increased threshold for taste and smell, together with chewing problems caused by teeth and muscle loss, can lead to the consumption of a restricted diet, poor in fibers and proteins that are known to strongly impact on microbiota composition[36,37]. Moreover, poor diet and diminished physical activity contribute to increase the chances of constipation and, consequently, of slower intestinal transit time, which may impact on the composition of the colonic microbiota due to the reduced bacterial excretion[48]. The age-related increased drug consumption[49] and the interaction between different medicines can also be listed among the possible factors that rule changes in the gut microbiota. The subject-specific combination of all these impacting environmental variables may be responsible for the inter-individual variability of the gut microbiota composition that is known to increase along with aging[50,51].
The aged-type gut microbiota is typically characterized by a reduced biodiversity, an increased abundance of opportunistic facultative anaerobes, and a decreased abundance of species with anti-inflammatory properties (i.e., Faecalibacterium prausnitzii and other butyrate producers)[7,44,52-55]. Interestingly, these deviations from the healthy adult-like profile overlap with those known to accompany several disorders characterized by systemic and/or chronic inflammation, such as obesity, metabolic syndrome and inflammatory bowel diseases[21,56,57]. Indeed, aging itself involves chronic immune and inflammatory unbalances. Elderly are generally affected by a process called “immunosenescence” that causes a decline in immune system functionality, and a chronic inflammatory status (“inflamm-aging”) characterizing the whole organism[58,59]. At the level of the gut, inflamm-aging could be responsible for an increased stimulation of the inflammatory response, allowing opportunistic pathogens (pathobionts) to thrive to the detriment of symbionts[60,61]. The age-related proliferation of opportunistic bacteria could both contribute to and be nurtured by inflamm-aging, in a sort of self-sustaining loop[55], possibly creating a predisposing environment for diseases the risk of which is known to increase along with age, such as colorectal cancer.
INTESTINAL MICROBIOTA AND COLORECTAL CANCER
Colorectal cancer (CRC) is the fourth most commonly diagnosed cancer in the Western world[62,63]. With more than one million of new cases and 600000 deaths per year, CRC undoubtedly constitutes a significant burden for public health in Western world.
CRC is the result of a multistep process whose progression is associated with the gradual accumulation of genetic and epigenetic mutations. Sporadic in more than 90% of the cases, CRC develops gradually, proceeding from normal epithelium to adenomatous polyps and invasive carcinoma, defining a process that can be slow, taking more than 10 years depending on the mutation frequency[64]. Several genetic predispositions which can increase cancer risk have been identified. The principal driver mutations involved in CRC include tumor suppressors adenomatous polyposis coli gene, β-catenin gene, deleted in colorectal cancer gene and p53[64], as well as the oncogenes Kirsten rat sarcoma[65] and myelocytomatosis oncogene[66,67]. However, even if within the last years a growing number of acquired genetic mutations have been described in CRC, trigger factors leading to their accumulation remain to be determined.
Environmental factors have been reported as the leading causes involved in CRC onset[68]. Chronic inflammation and diet have been historically recognized as the prominent CRC drivers[69,70], however, recently, a new potential factor in CRC is emerging: the human intestinal microbiota[71-73]. For instance, the relevance of a compromised microbiota-host homeostasis in CRC onset has been highlighted by the recent finding that mice defective in the inflammasome function have an increased risk to develop CRC[74]. While the involvement of diet and inflammation in CRC has been proved by “traditional” observational and epidemiological studies[70,75-77], only the recent widespread of next-generation sequencing (NGS)-based approaches for gut microbiota characterization allowed to identify characteristic ecosystem changes associated with CRC. Comparative NGS studies of the gut microbiota structure in stools, luminal samples and swabs from CRC patients and age-matched healthy controls have been carried out[78-80]. With respect to healthy controls, CRC patients were significantly enriched in fecal Fusobacterium, Enterococcaceae, Campylobacter, Erysipelotrichaceae, Collinsella, Peptostreptococcus and Anaerotruncus, and depleted in members of the Clostridium cluster IV, such as Faecalibacterium prausnitzii (F. prausnitzii) and Roseburia. On the intestinal mucosa, CRC patients showed an increase of Porphyromonas, Fusobacterium, Peptostreptococcus and Mogibacterium, whereas Faecalibacterium, Blautia and Bifidobacterium were depleted. This CRC-associated microbiome is enriched in pro-inflammatory opportunistic pathogens, e.g., Fusobacterium, Enterococcaceae and Campylobacter[81-85], and microorganisms commonly associated with metabolic disorders, such as Erysipelotrichaceae[86,87], while depleted in microbial partners strategic to preserve the intestinal homeostasis[88], such as well-known butyrate producers (i.e., F. prausnitzii and Roseburia) and protective bifidobacteria[22,89]. These NGS data reflect an overall pro-inflammatory configuration for the CRC-associated gut microbial ecosystem, which can concur in compromising the microbiota-host mutualism and, eventually, consolidate the disease state. Very recently, comparative analyses of mucosal microorganisms on cancerous tissue and matched non-cancerous tissue have been carried out, allowing to detect microorganisms specifically enriched on CRC tumor sites[79,84,85]. Cancerous mucosa showed an overall decrease in bacterial diversity with respect to non-cancerous tissues, and was characterized by a reduction in Faecalibacterium and higher abundances of Fusobacterium, Bacilli and Phascolarctobacterium. These pro-inflammatory microorganisms can modulate the tumor microenvironment, affecting the course of CRC progression.
In order to explore dysbiosis of the gut microbiota in CRC at the community level, we sought associations between individual genera. To this aim, we obtained co-abundance groups (CAGs), groups of microorganisms which correlate and cluster together, by a bioinformatics analysis[90] of the publicly available dataset from Wu et al[80], a well characterized case-control study of the CRC-associated microbiome. Six CAGs displaying significantly different inter-relationships from each other (P < 0.001) have been identified: Fusobacterium CAG, Prevotella CAG, Barnesiella CAG, Coprobacillus CAG, Faecalibacterium CAG and Bifidobacterium CAG. Significant associations between bacterial genera have been calculated and represented in a Wiggum plot (Figure 1). This network analysis allowed us to describe - to our knowledge for the first time - microbial co-abundance networks which include microorganisms previously associated with CRC risk or protection. According to our analysis, the CRC-associated microorganisms Fusobacterium and Erysipelotrichaceae belong to the same CAG (Fusobacterium). Analogously, CRC-associated groups as Enterobacteriaceae, Escherichia, Shigella and Klebsiella co-vary within the same cluster (Prevotella). On the other hand, a common CAG (Bifidobacterium) is shared by non-CRC-associated groups as Bifidobacterium and Lachnospiraceae (a family member of the Clostridium cluster IV). Other health-promoting mutualists belonging to the Clostridium cluster IV, such as Faecalibacterim, Blautia, Roseburia, Dorea and Lachnospiraceae, group together in Faecalibacterim CAG. Finally, we identified one CAG (Barnesiella) including both pro-carcinogenic microorganisms as Porphyromonadaceae and Eubacterium, as well as protective members of the Clostridium cluster IV (Ruminococcus, Butyrococcus and Oscillibacter). Even if data from this computational analysis must be taken with caution since based on a limited dataset, we can hypothesize the existence of 3 pro-carcinogenic CAGs (Fusobacterium CAG, Prevotella CAG and Coprobacillus CAG) and 2 CRC protective CAGs (Bifidobacterium CAG and Faecalibacterium CAG).
Figure 1.

Network plot showing correlation relationships among clusters of bacterial genera for the Wu et al[80] dataset. Each node represents a taxon color-coded for co-abundance group and each line highlights a significant correlation between two bacterial genera. Circle size is proportional to genus abundance. Solid lines indicate positive correlation, whereas dot lines indicate negative correlation. Thickness of the lines is proportional to correlation strength.
Suggesting the involvement of specific microbiota dysbiosis in CRC, NGS-based microbiome studies are imposing a more holistic vision of the interplay between environment and genetics in CRC, where dietary factors and inflammation need to be considered against the background in the microbiota-host interaction process (Figure 2). However, the static nature of these studies did not permit to comprehend whether dysbiosis are a cause or a consequence of the disease onset. Further, these descriptive studies did not provide information on either the mechanisms by which members of the gut microbial ecosystem can influence the CRC, or, more importantly, the triggers that shift the microbiota towards a carcinogenic configuration. With the attempt to deal with these questions, a new approach to study the role of microorganisms in CRC onset is emerging. Pairing NGS-based microbiota surveys and the usage of germ-free (GF), conventionalized and mono associated mice to test mechanistic hypotheses, new insights on the microbial ecology of CRC have been provided[73].
Figure 2.
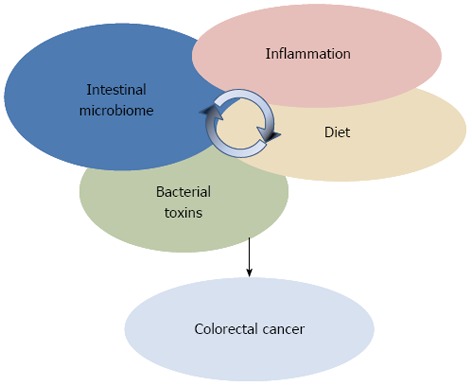
Colorectal cancer arises from the interplay between endogenous and exogenous factors, such as inflammation, diet, intestinal microbiome structure, and transcription and activity of bacterial genotoxins.
Bacterial driver-passenger model
Recently, a first dynamic model of the microbial ecology involved in CRC onset and progression has been proposed by Tjalsma et al[73]: the bacterial driver-passenger model. According to this model, CRC development is initiated by indigenous bacteria with pro-carcinogenic features - defined as bacterial drivers - that drive epithelial DNA damage and contribute to CRC initiation. In a subsequent step, the local microenvironment is altered as a consequence of the ongoing tumorigenesis and bacterial drivers are replaced by bacterial passengers, microorganisms showing a competitive advantage in the tumor microenvironment and being capable of nurturing tumor progression. For instance, nutrients and co-factors specific of the tumor microenvironment - such as the presence of reactive oxygen species - can be selectively utilized by specific bacterial passengers[91].
Bacterial drivers are defined as intestinal bacteria showing pro-carcinogenic features - either transient or autochthonous microbiota components - that may initiate the process of carcinogenesis. Several candidate bacterial drivers have been identified (Table 1), such as superoxide-producing strains of Enterococcus faecalis[92], genotoxin-producing Escherichia coli strains[93], and toxigenic strains of Bacteroides fragilis[94]. Furthermore, pro-inflammatory members of Enterobacteriaceae, such as Shigella, Citrobacter and Salmonella have been associated with early stages of CRC as possible bacterial drivers[95,96]. Occasionally, bacterial drivers act in concert with helper bacteria (or α-bugs) in carcinogenesis promotion[97]. Generally belonging to pro-inflammatory Enterobacteriaceae, these microorganisms are proposed to crowd out symbiont CRC-protecting anti-inflammatory microbiota components, such as F. prausnitzii, Roseburia or Bifidobacterium, favoring the subsequent tissue colonization by drivers.
Table 1.
Microorganisms involved in colorectal cancer
| Microorganism | Role in CRC | Mechanism | Ref. |
| E. faecalis | Driver | Production of superoxide | [92] |
| E. coli NC101 | Driver | Genotoxin production (colibactin) | [122] |
| B. fragilis | Driver | Genotoxin production (fragilisin) | [94] |
| Shigella | Driver | Induction of inflammation | [73] |
| Citrobacter | Driver | Induction of inflammation | [73] |
| Salmonella | Driver | Induction of inflammation | [73] |
| Enterobacteriaceae | Helper | Induction of inflammation | [73] |
| Fusobacterium | Passenger | Induction of inflammation | [84] |
| S. gallolyticus | Passenger | Induction of inflammation | [98] |
| C. septicum | Passenger | Induction of inflammation | [99] |
| F. prausnitzii | Protective | Butyrate production; anti-inflammatory properties | [78] |
| Roseburia | Protective | Butyrate production; anti-inflammatory properties | [78] |
| Bifidobacterium | Protective | Protection from pathogens; anti-inflammatory properties | [71] |
| Corynebacteriaceae | Protective | Anti-inflammatory properties | [78] |
Microorganisms involved in colorectal cancer (CRC), their role as driver, passenger or protective bacteria and the mechanisms involved in CRC induction or protection. E. faecalis: Enterococcus faecalis; E. coli: Escherichia coli; B. fragilis: Bacteroides fragilis; S. gallolyticus: Streptococcus gallolyticus; C. septicum: Clostridium septicum; F. prausnitzii: Faecalibacterium prausnitzii.
Passenger bacteria are always autochthonous members of the gut microbial community. Relatively poor colonizer of a healthy intestinal tract, passengers show a competitive advantage in the tumor microenvironment (Table 1). However, differently from drivers, which are always pro-carcinogenic, passenger bacteria can be of either pro-carcinogenic or protective nature, depending on the microorganism. While in some cases the carcinogenic tissue has been shown to be selectively colonized by opportunistic pathogens, such as Fusobacterium[78,83,84], Streptococcus gallolyticus[98] and Clostridium septicum[99], which can be involved in CRC progression, in other circumstances the tumor sites were enriched in passenger bacteria belonging to well-known mutualistic microbiota components, as Corynebacteriaceae, Roseburia and Faecalibacterium, suggesting a possible protective role for these microorganisms as CRC quencher[78].
Mechanisms possibly involved in microbial CRC promotion
Gut microorganisms may promote CRC onset and progression by different processes (Table 1)[71], such as (1) the induction of a chronic inflammatory state; (2) the biosynthesis of genotoxins interfering with the cell cycle regulation or directly damaging DNA; (3) the production of toxic metabolites; and (4) the activation of dietary heterocyclic amines to pro-carcinogenic compounds. Here we will specifically discuss the role of three of these factors - inflammation, genotoxins and toxic metabolites – in CRC onset and progression.
Chronic inflammatory disorders are associated with a higher risk of cancer development[100]. Inflammation can nurture carcinogenesis by inducing gene mutations, inhibiting apoptosis or stimulating angiogenesis and cell proliferation. By regulating cell survival, inflammation and immunity, nuclear factor (NF)-κB is at the connection between inflammation and cancer. In particular, experiments carried out in mouse models of colitis-associated cancer have been successful in demonstrating a dual role for NF-κB in carcinogenesis, which depends on the cell type[101]. While in enterocytes NF-κB contributes to tumor initiation by suppressing apoptosis, in myeloid cells it is involved in the promotion of tumor growth by means of the production of inflammatory mediators. Further, it has been recently demonstrated that elevated NF-κB signaling can activate mutations in the Wnt pathway, leading to the differentiation of epithelial non-stem cells into tumor-initiating cells[102]. Generally, the activation of NF-kB results in the expression of inflammatory cytokines [e.g., tumour necrosis factor-alpha, interleukin (IL)-1, IL-6 and IL-8), adhesion molecules, enzymes involved in prostaglandin synthesis, nitric oxide synthase, angiogenic factors and anti-apoptotic genes, providing survival advantages to precancerous or tumor cells in the gut[75,103]. The activation of NF-κB as a result of microbial sensing via the host Toll-like receptors (TLRs) has been proposed to support intestinal tumor growth under steady-state conditions[104,105]. Several evidences have been reported in support of the role of the gut microbiota in the inflammation-dependent carcinogenesis in the gut. Crohn’s disease and ulcerative colitis are often associated with an increased risk of developing CRC and epidemiological data suggest that duration and severity of chronic colitis represent a significant risk factor for colitis-associated CRC[106,107]. Furthermore, microbiota unbalances in favor of pro-inflammatory opportunistic pathogens as Enterobacteriaceae and Clostridium difficile have been indicated to be involved in tumor progression[108,109] and, in the context of the bacterial driver-passenger model, several bacterial drivers, such as Shigella, Citrobacter, Salmonella and toxigenic Bacteroides fragilis (B. fragilis), as well as the passengers Fusobacterium and Streptococcus gallolyticus and Clostridium septicum, have been reported to support carcinogenesis by the induction of a pro-inflammatory response[73]. Strikingly, by inducing azoxymethane (AOM)-colitis in conventional and GF IL-10 knockout (Il10-/-) mice, Uronis et al[110] were successful in demonstrating that microbial sensing via TLRs is essential to develop colitis-associated CRC.
Inflammation also represents a molecular link between host immune response, intestinal microbiota and genotoxic events in the inflammation-associated CRC[111]. Several bacterial taxa that belong to the human gut microbiome in a subset of the healthy population contain toxin-producing strains[5]. The long-term effects of chronic exposure to low doses of such bacteria as well as the eventual contribution to the carcinogenic process of bacterial toxins remain to be elucidated. Toxins impinge on key eukaryotic processes, such as cellular signaling, and some directly attack the genome[112] these last by damaging DNA, either directly, by enzymatic attack, or indirectly, by provoking an inflammatory reaction that produces free radicals. Also, they can affect DNA repair mechanisms.
The capacity of the B. fragilis toxin (BFT)-producing strains to promote colon tumorigenesis is mediated by the increased expression of STAT3 that leads to the recruitment of the highly pro-inflammatory subset of T helper type 17 lymphocytes, suggesting that the pro-carcinogenic role of BFT is to promote a de-regulated inflammatory response[113]. BFT is a metalloprotease known to bind to colonic epithelial cells and stimulate cleavage of E-cadherin, thus increasing intestinal barrier permeability and augmenting cell signalling via the β-catenin/Wnt pathway, which is constitutively activated in essentially all CRC. As a result, BFT stimulates proliferation and migration of human colon cancer cells in vitro[114]. It is worth noting that the enterotoxigenic form of B. fragilis (ETBF) is only present in approximately 10%-20% of the healthy population whereas the fecal carriage of ETBF is increased of about 40% in CRC patients[94,115].
However, although the B. fragilis toxin has been proposed as one of the main CRC driving suspects on the basis of experimental work[113,116], very recent studies show that the most actively transcribed toxins in tumor tissue and surrounding mucosa from CRC patients are those derived from Escherichia coli (E. coli), Salmonella enterica and Shigella flexneri. This suggests a strong involvement of enterobacterial toxins in tumorigenesis. Also in this context, inflammation has been shown to increase toxigenic E. coli strains, promoting their adhesion to the host epithelia[111]. A number of E. coli strains produce a wide array of toxins, some of which are turning out to be potentially harmful in humans, either directly damaging DNA or specifically disrupting cell signaling.
The cytolethal distending toxins (CDTs), which comprise a family of intracellular-acting bacterial protein toxins produced by several gram-negative bacteria, belong to the first group. Their activity upon eukaryotic cells results in several consequences, the most characteristic of which is the induction of G(2)/M cell cycle arrest[117]. Active CDTs consist of three subunits: CdtA and CdtC, which guide internalization, and CdtB, which enzymatically induces DNA double-strand breaks that recruit and activate the ataxia telangiectasia mutated kinase, thus triggering a DNA damage response (DDR). The DDR provides an efficient barrier to tumorigenesis through induction of cell death or senescence[118]. Cells exposed to sub-lethal doses of the CDTs from Helicobacter hepaticus (H. hepaticus) or Haemophilus ducreyi exhibit increased frequency of mutations, accumulation of chromosomal aberrations and enhanced anchorage-independent growth[119]. Furthermore, chronic infection of mouse liver and intestine with CDT-producing H. hepaticus or Campylobacter jejuni, respectively, is associated with dysplasia[119], confirming the capacity of CDT-producing bacteria to induce pre-neoplastic lesions in vivo. Very recently, Buc et al[120] demonstrated a high prevalence of genotoxin- and cyclomodulin-producing mucosa-associated E. coli strains in CRC patients.
Furthermore, some commensal E. coli strains of the phylogenetic group B2 harbour a 54 kb polyketide synthase (pks) pathogenicity island encoding the enzymes required for the synthesis of a putative hybrid peptide-polyketide genotoxin, named colibactin[121]. Infection of mice with a pks+ E. coli strain has been linked to the expression of pks genes required for colibactin production as well as to DNA damage induction[122]. The capacity of colibactin to promote tumorigenesis in vivo has been recently proven in an animal model of colitis-associated CRC. GF IL-10 knockout mice treated with the colon-specific carcinogen AOM and monocolonized with pks+ E. coli showed a high incidence of invasive adenocarcinoma if compared to mice infected with an isogenic pks-deficient strain or the control commensal bacterium pks-E. faecalis[93]. The detection of E. coli isolates carrying the pks island in 66.7% CRC patients compared to 20% found in non-inflammatory bowel disease/non-CRC controls suggests a concerted action of host inflammation and E. coli-derived pks in giving rise to a host microenvironment that promotes DNA damage and tumorigenesis[93]. These authors also showed that optimal colonization by colibactin-producing E. coli strains is established in an already-inflamed gut. In fact, by remodeling the intestinal immune response and shifting the colonic bacterial community to one that further promotes CRC, bacterial drivers permit the colonization of colibactin-producing E. coli strains that actively contribute to disease progression.
A second group of toxins includes those disrupting the cell signaling that regulates cell proliferation or induces inflammation. The E. coli cytotoxic necrotizing factor 1 (CNF1), which is expressed by many human isolates, activates the Rho GTPases[123], inducing dysfunctions in already transformed epithelial cells, such as apoptosis counteraction, pro-inflammatory cytokines’ release, COX2 expression, NF-κB activation and boosted cellular motility. Also, CNF1 induces quiescent cells to enter the cell cycle and undergo DNA synthesis[124], interferes with normal cytokinesis, resulting in the production of multinucleated cells and in the onset of aneuploidia. As cancer may arise when the same regulatory pathways are affected, it is conceivable that CNF1-producing E. coli infections can contribute to cancer development[125]. Our hypothesis is that these bacteria may act as passengers, reinforcing and favoring but not causing the development of colorectal cancer. The pro-inflammatory capacity of CNF1 has recently been confirmed in Drosophila, where the toxin could activate one of the key transcription factors of the innate immune response, namely NF-κB, independently of the triggering of pathogen recognition receptors. Indeed, the CNF1-mediated activation of the Rac2 GTPase triggers protective immunity via the innate Rip kinase signaling that functions upstream of NF-κB[126]. Taken altogether, these data support the strategic role of toxigenic E. coli strains in CRC onset and progression.
The gut microbiome is a major driver in shaping the gut metabolome[127]. Among microbial metabolites, several have been identified as potentially important carcinogens or protective. Secondary bile acids in particular have been detected in elevated levels in CRC patient stools and have been shown to have carcinogenic properties in vitro[128]. A long list of other metabolites are suspected at varying degrees to be implicated in CRC development, such as hydrogen sulfide[129], proteolysis products (ammonia, amines, phenols)[130], and acetaldehyde[131]. Butyrate is the most sought-after beneficial metabolite as it is a major energy source for colonocytes and more importantly has an anti-proliferative activity and induces apoptosis of CRC cells in vitro[132].
Triggering factors that force microbiota to become carcinogenic
Besides its role in CRC onset, inflammation surely exerts a central role in triggering the carcinogenic potential of the gut microbial ecosystem (Figure 3)[93]. Experiments relying on mice defective in components of the immune system successfully demonstrated that chronic inflammation alters the intestinal microbial community composition towards a configuration that predisposes to the disease[133]. According to Garrett et al[134], Tbet-/-/Rag2-/- mice, which are deficient in adaptive and innate immune function, developed a colitis phenotype transmissible to wild-type mice by the adoptive transfer of their gut microbiota. Analogously, mice lacking the bacterial flagellin receptor TLR5 exhibited a syndrome encompassing insulin resistance, hyperlipidemia, and increased fat deposition associated with microbiota alterations. Strikingly, these metabolic changes were transferable to wild-type mice by acquiring the Tlr5-/- gut microbiota[134]. In this context, Arthur et al[93] specifically demonstrated that intestinal inflammation can boost the cancer-inducing activity of the gut microbiota. According to the Authors, chronic inflammation in Il10-/- mice was sufficient to prompt microbiota shifts, supporting the AOM-induced carcinogenesis. Favoring the adhesion of driver bacteria with genotoxic potential to the colonic mucosa - as well as the overall expansion of pro-inflammatory Enterobacteriaceae in the gut - inflammation creates the environment that supports a bacteria-mediated carcinogenesis process. In particular, Arthur et al[93] showed that chronic colitis in Il10-/- mice was sufficient to favor a dramatic expansion of E. coli NC101 on the intestinal mucosa. Harboring a pks pathogenicity island, E. coli NC101 codes for the genotoxin colibactin[121] that allows this microorganism to accelerate progression from dysplasia to invasive carcinoma. Inflammation in the gut is also pivotal to initiate a microbiota-dependent pro-inflammatory loop detrimental for host health[71]. An aberrant inflammatory response in the gut can shift the balance between protective mutualists and pathobionts in favor of the latter[135,136]. By inducing a pro-inflammatory loop, these microorganisms can work as bacterial drivers, consolidating the inflammatory state[28] and resulting in a self-sustained pro-inflammatory response that affects the microbial ecology of the human gut, further compromising the microbiota-host mutualism and supporting CRC.
Figure 3.

Environmental triggers, such as diet, aging and pathogen infections, can force microbiota changings that, in a genetically susceptible host, can drive to chronic inflammation in the gut. Inflammation shifts the gut microbiota towards a pro-inflammatory configuration, supporting colorectal cancer (CRC) drivers as pathobionts at the expense of health-promoting CRC-protective microbiota components. As a consequence, a pro-inflammatory loop is established in the gut, directly supporting CRC onset and favoring colonization by toxigenic bacterial drivers directly involved in CRC promotion.
Abnormal dietary inputs can lead to the expansion of pro-inflammatory microbes in the gut[137]. For instance, a diet rich in saturated milk fat has been reported to induce the expansion of Bilophila wadsworthia, which may favor carcinogenesis in the gut by promoting pro-inflammatory TH1[138]. Indeed, high-fat diet impacts on gut microbiome have seen increased interest in the recent years as fat has been linked epidemiologically to intestinal inflammation and diseases. While as expected a high-fat diet modifies the microbiome, the fact that different fat compositions induced different changes in animal models calls for a more controlled dietary intervention in humans. For example, observational data suggested that Western diet (protein- and fat-enriched) and African diet (polysaccharide-enriched) drive strikingly different microbiomes, possibly explaining different CRC rates[13,38,139]. Reciprocal diet exchanges indeed demonstrated that the microbiome and metabolome were rapidly responsive towards respective “beneficial” and “detrimental” states, as well as markers of mucosal proliferation[13].
The process of human aging has a well-documented impact on the gut microbiota structure[48,140], raising the question of whether age-related microbiota dybioses can trigger a microbiota-dependent carcinogenic process in the gut. Showing a pro-inflammatory configuration, the aged-type gut microbial ecosystem can force a microbiota-dependent pro-inflammatory loop in the gut, compromising the microbiota-host mutualism and supporting carcinogenesis. Strengthening this hypothesis, the incidence of CRC has been reported to increase in the elderly; about 50% of the Western population develops colorectal polyps at the age of 70 and 5% of these polyps progress to cancer[141].
The pervasive role of genotoxins in CRC onset and progression, led researchers to investigate triggering factors that govern toxin biosynthesis and activity. Environmental changes in the gut ecosystem, such as changes in pH, in oxygen availability or the presence of a specific metabolite, have been suggested to have a role in the modulation of toxin transcription. Intriguingly, interspecies quorum sensing resulting from microbe/microbe interaction processes has been suggested to play a role in governing bacterial toxin production in the gut[72,142]. Even if research in this field is still in its infancy, recent experimental works demonstrated the strategic role of microbe/microbe, microbe/host and microbe/environment interaction processes in regulating bacterial virulence and toxin activity. In a recent experimental research based on GF and conventional mice, Kamada et al[143] demonstrated that changes in dietary substrates can result in a microbiota-dependent regulation of virulence factors. According to the Authors, dietary changes can boost commensals capable to outcompete toxigenic pathogens for food sources, resulting in the down-regulation of virulence genes and eventually pathogen clearance. Further, Marks et al[144] demonstrated that interkingdom signaling as a result of the host response to the influenza A virus infection was sufficient to trigger the expression of Streptococcus pneumoniae virulence genes, resulting in the transition from commensalism to pathogenicity. Even if S. pneumoniae is a common human nasopharyngeal opportunistic bacterium, these findings allow us to hypothesize the existence of analogous processes in the gut ecosystem, resulting in the activation of a virulence phenotype and toxin transcription of enterotoxigenic CRC drivers.
CONCLUSION
The worldwide diffusion of NGS-based microbiota surveys in CRC patients, alongside the utilization of GF, mono associated and humanized mice, led to an increasing perception of the pivotal role exerted by the gut microbiota in CRC onset and progression. Lights on the microbial ecology of the process have been provided, and possible mechanisms involved suggested. This brought the researchers to focus their attention on triggering factors that turn the intestinal microbiota from a mutualistic configuration to a CRC-promoting asset. Inflammation has undoubtedly a central role in this process, being a common outcome shared by different triggering factors, such as diet, aging, microbe-microbe and microbe-host interactions (Figure 3). In fact, changes in diet, aging, as well as pathobiont-dependent pro-inflammatory dysbiosis of the gut microbiota, can force the gut microbiota to a pro-inflammatory asset, changing the microecology of the gut ecosystem and activating toxigenic CRC bacterial drivers. In this context, of extraordinary importance will be the development of strategies able to interfere and/or block these triggering factors, preserving the microbiota-host mutualism along the entire life span. Different approaches can be implemented. Since diet represents the pivotal strategy to modulate composition and functionality of the gut microbiota, the most promising approach to preserve microbiota-host mutualisms relies on dietary interventions. For instance, diet can be modulated to boost health-promoting microbiota groups, such as anti-inflammatory members of the Clostridium cluster IV or short chain fatty acid producers of the Clostridium cluster XIVa. Strengthening this perspective, in a life-long longitudinal study carried out in mice, Zhang et al[145] demonstrated that different diets modulated differently the microbiome trajectories along with aging. In particular, according to the Authors low-fat diet and caloric restriction increased the relative abundance of phylotypes positively associated with the life span in the middle-life, and, at the same time, lowered the abundance of opportunistic pro-inflammatory pathogens, which could represent CRC bacterial drivers.
A second approach for CRC prevention surely relies on the usage of probiotic bacteria, such as Bifidobacterium and Lactobacillus. Probiotics have been demonstrated to be effective in reducing CRC risk in humans[146-149]. Showing immunomodulating properties, antimicrobial activities, as well as the capacity to interfere with toxin synthesis and activity, probiotic bacteria can act simultaneously on different CRC triggering factors. In fact, probiotics have been reported as effective in quenching host inflammatory response[150], in inhibiting the colonization of known CRC drivers[22,151] and in inactivating bacterial toxins[152] or interfering with their production[153,154].
Even if significant steps forward have been carried out, we are still far from fully appreciating the multifactorial role of the intestinal microbiota in CRC. More longitudinal microbiome surveys need to be carried out, and intestinal polyps as well as adenocarcinoma tissues must be sampled, in order to follow the gut microbiota dynamics over time for the development of colonic neoplasia. Microbiota on tumor sites needs to be compared with off-tumor matched tissues, as a better comparison than mucosal samples from healthy patients. Associations between structure and dynamics of the gut microbiome and the different stages of colonic neoplasia need to be better defined, causality should be further explored, possibly by using GF, mono associated as well as humanized animal models. Finally, meta-analysis integrating epidemiological studies with microbiome datasets will allow us to better define triggering factors that force the microbiota to become carcinogenic, so that hypotheses can be verified in mice where possible intervention strategies can be tested.
Footnotes
P- Reviewers: Abdel-Salam OME, Beyaert R, Cairo G, Teramoto-Matsubara OT S- Editor: Gou SX L- Editor: A E- Editor: Wu HL
References
- 1.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dethlefsen L, Eckburg PB, Bik EM, Relman DA. Assembly of the human intestinal microbiota. Trends Ecol Evol. 2006;21:517–523. doi: 10.1016/j.tree.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 3.O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7:688–693. doi: 10.1038/sj.embor.7400731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajilić-Stojanović M, Smidt H, de Vos WM. Diversity of the human gastrointestinal tract microbiota revisited. Environ Microbiol. 2007;9:2125–2136. doi: 10.1111/j.1462-2920.2007.01369.x. [DOI] [PubMed] [Google Scholar]
- 7.Rajilić-Stojanović M, Heilig HG, Molenaar D, Kajander K, Surakka A, Smidt H, de Vos WM. Development and application of the human intestinal tract chip, a phylogenetic microarray: analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ Microbiol. 2009;11:1736–1751. doi: 10.1111/j.1462-2920.2009.01900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, Ugarte E, Muñoz-Tamayo R, Paslier DL, Nalin R, et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol. 2009;11:2574–2584. doi: 10.1111/j.1462-2920.2009.01982.x. [DOI] [PubMed] [Google Scholar]
- 9.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turnbaugh PJ, Quince C, Faith JJ, McHardy AC, Yatsunenko T, Niazi F, Affourtit J, Egholm M, Henrissat B, Knight R, et al. Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc Natl Acad Sci USA. 2010;107:7503–7508. doi: 10.1073/pnas.1002355107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA. 2010;107:18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Candela M, Biagi E, Maccaferri S, Turroni S, Brigidi P. Intestinal microbiota is a plastic factor responding to environmental changes. Trends Microbiol. 2012;20:385–391. doi: 10.1016/j.tim.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Ou J, Carbonero F, Zoetendal EG, DeLany JP, Wang M, Newton K, Gaskins HR, O’Keefe SJ. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr. 2013;98:111–120. doi: 10.3945/ajcn.112.056689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeffery IB, Claesson MJ, O’Toole PW, Shanahan F. Categorization of the gut microbiota: enterotypes or gradients? Nat Rev Microbiol. 2012;10:591–592. doi: 10.1038/nrmicro2859. [DOI] [PubMed] [Google Scholar]
- 16.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A, Waller A, Mende DR, Kultima JR, Martin J, et al. Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol. 2012;10:323–335. doi: 10.1038/nrmicro2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology. 2009;136:65–80. doi: 10.1053/j.gastro.2008.10.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469:543–547. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
- 23.Corr SC, Li Y, Riedel CU, O’Toole PW, Hill C, Gahan CG. Bacteriocin production as a mechanism for the antiinfective activity of Lactobacillus salivarius UCC118. Proc Natl Acad Sci USA. 2007;104:7617–7621. doi: 10.1073/pnas.0700440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keeney KM, Finlay BB. Enteric pathogen exploitation of the microbiota-generated nutrient environment of the gut. Curr Opin Microbiol. 2011;14:92–98. doi: 10.1016/j.mib.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stecher B, Hardt WD. Mechanisms controlling pathogen colonization of the gut. Curr Opin Microbiol. 2011;14:82–91. doi: 10.1016/j.mib.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. 2012;489:231–241. doi: 10.1038/nature11551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Candela M, Biagi E, Turroni S, Maccaferri S, Figini P, Brigidi P. Candela M, Biagi E, Turroni S, Maccaferri S, Figini P, Brigidi P. Dynamic efficiency of the human intestinal microbiota. Crit Rev Microbiol. 2013:In press. doi: 10.3109/1040841X.2013.813900. [DOI] [PubMed] [Google Scholar]
- 28.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collins SM, Bercik P. The relationship between intestinal microbiota and the central nervous system in normal gastrointestinal function and disease. Gastroenterology. 2009;136:2003–2014. doi: 10.1053/j.gastro.2009.01.075. [DOI] [PubMed] [Google Scholar]
- 30.Rhee SH, Pothoulakis C, Mayer EA. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat Rev Gastroenterol Hepatol. 2009;6:306–314. doi: 10.1038/nrgastro.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13:701–712. doi: 10.1038/nrn3346. [DOI] [PubMed] [Google Scholar]
- 32.Collins SM, Bercik P. Gut microbiota: Intestinal bacteria influence brain activity in healthy humans. Nat Rev Gastroenterol Hepatol. 2013;10:326–327. doi: 10.1038/nrgastro.2013.76. [DOI] [PubMed] [Google Scholar]
- 33.Zoetendal EG, Akkermans AD, De Vos WM. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl Environ Microbiol. 1998;64:3854–3859. doi: 10.1128/aem.64.10.3854-3859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guarner F, Malagelada JR. Gut flora in health and disease. Lancet. 2003;361:512–519. doi: 10.1016/S0140-6736(03)12489-0. [DOI] [PubMed] [Google Scholar]
- 35.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, et al. The long-term stability of the human gut microbiota. Science. 2013;341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5:220–230. doi: 10.1038/ismej.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jalanka-Tuovinen J, Salonen A, Nikkilä J, Immonen O, Kekkonen R, Lahti L, Palva A, de Vos WM. Intestinal microbiota in healthy adults: temporal analysis reveals individual and common core and relation to intestinal symptoms. PLoS One. 2011;6:e23035. doi: 10.1371/journal.pone.0023035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ehlers S, Kaufmann SH. Infection, inflammation, and chronic diseases: consequences of a modern lifestyle. Trends Immunol. 2010;31:184–190. doi: 10.1016/j.it.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 42.Ohnmacht C, Marques R, Presley L, Sawa S, Lochner M, Eberl G. Intestinal microbiota, evolution of the immune system and the bad reputation of pro-inflammatory immunity. Cell Microbiol. 2011;13:653–659. doi: 10.1111/j.1462-5822.2011.01577.x. [DOI] [PubMed] [Google Scholar]
- 43.Jernberg C, Löfmark S, Edlund C, Jansson JK. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology. 2010;156:3216–3223. doi: 10.1099/mic.0.040618-0. [DOI] [PubMed] [Google Scholar]
- 44.Mäkivuokko H, Tiihonen K, Tynkkynen S, Paulin L, Rautonen N. The effect of age and non-steroidal anti-inflammatory drugs on human intestinal microbiota composition. Br J Nutr. 2010;103:227–234. doi: 10.1017/S0007114509991553. [DOI] [PubMed] [Google Scholar]
- 45.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viswanathan V. The meddling microbes midst our medicines. Gut Microbes. 2013:Epub ahead of print. doi: 10.4161/gmic.26250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Bäckhed HK, Gonzalez A, Werner JJ, Angenent LT, Knight R, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150:470–480. doi: 10.1016/j.cell.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biagi E, Candela M, Fairweather-Tait S, Franceschi C, Brigidi P. Aging of the human metaorganism: the microbial counterpart. Age (Dordr) 2012;34:247–267. doi: 10.1007/s11357-011-9217-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Genser D. Food and drug interaction: consequences for the nutrition/health status. Ann Nutr Metab. 2008;52 Suppl 1:29–32. doi: 10.1159/000115345. [DOI] [PubMed] [Google Scholar]
- 50.Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4586–4591. doi: 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rea MC, O’Sullivan O, Shanahan F, O’Toole PW, Stanton C, Ross RP, Hill C. Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J Clin Microbiol. 2012;50:867–875. doi: 10.1128/JCM.05176-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayashi H, Sakamoto M, Kitahara M, Benno Y. Molecular analysis of fecal microbiota in elderly individuals using 16S rDNA library and T-RFLP. Microbiol Immunol. 2003;47:557–570. doi: 10.1111/j.1348-0421.2003.tb03418.x. [DOI] [PubMed] [Google Scholar]
- 53.Mueller S, Saunier K, Hanisch C, Norin E, Alm L, Midtvedt T, Cresci A, Silvi S, Orpianesi C, Verdenelli MC, et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: a cross-sectional study. Appl Environ Microbiol. 2006;72:1027–1033. doi: 10.1128/AEM.72.2.1027-1033.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mariat D, Firmesse O, Levenez F, Guimarăes V, Sokol H, Doré J, Corthier G, Furet JP. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009;9:123. doi: 10.1186/1471-2180-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Biagi E, Nylund L, Candela M, Ostan R, Bucci L, Pini E, Nikkïla J, Monti D, Satokari R, Franceschi C, et al. Through ageing, and beyond: gut microbiota and inflammatory status in seniors and centenarians. PLoS One. 2010;5:e10667. doi: 10.1371/journal.pone.0010667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cani PD, Delzenne NM. The role of the gut microbiota in energy metabolism and metabolic disease. Curr Pharm Des. 2009;15:1546–1558. doi: 10.2174/138161209788168164. [DOI] [PubMed] [Google Scholar]
- 57.Maslowski KM, Mackay CR. Diet, gut microbiota and immune responses. Nat Immunol. 2011;12:5–9. doi: 10.1038/ni0111-5. [DOI] [PubMed] [Google Scholar]
- 58.Ostan R, Bucci L, Capri M, Salvioli S, Scurti M, Pini E, Monti D, Franceschi C. Immunosenescence and immunogenetics of human longevity. Neuroimmunomodulation. 2008;15:224–240. doi: 10.1159/000156466. [DOI] [PubMed] [Google Scholar]
- 59.Larbi A, Franceschi C, Mazzatti D, Solana R, Wikby A, Pawelec G. Aging of the immune system as a prognostic factor for human longevity. Physiology (Bethesda) 2008;23:64–74. doi: 10.1152/physiol.00040.2007. [DOI] [PubMed] [Google Scholar]
- 60.Guigoz Y, Doré J, Schiffrin EJ. The inflammatory status of old age can be nurtured from the intestinal environment. Curr Opin Clin Nutr Metab Care. 2008;11:13–20. doi: 10.1097/MCO.0b013e3282f2bfdf. [DOI] [PubMed] [Google Scholar]
- 61.Sansonetti PJ. To be or not to be a pathogen: that is the mucosally relevant question. Mucosal Immunol. 2011;4:8–14. doi: 10.1038/mi.2010.77. [DOI] [PubMed] [Google Scholar]
- 62.Boyle P, Ferlay J. Cancer incidence and mortality in Europe, 2004. Ann Oncol. 2005;16:481–488. doi: 10.1093/annonc/mdi098. [DOI] [PubMed] [Google Scholar]
- 63.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 64.Vogelstein B, Kinzler KW. Colorectal tumors. The Genetic Basis of Human Cancer. New York: McGraw-Hill; 1998. pp. 565–587. [Google Scholar]
- 65.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 66.Sheng H, Shao J, Williams CS, Pereira MA, Taketo MM, Oshima M, Reynolds AB, Washington MK, DuBois RN, Beauchamp RD. Nuclear translocation of beta-catenin in hereditary and carcinogen-induced intestinal adenomas. Carcinogenesis. 1998;19:543–549. doi: 10.1093/carcin/19.4.543. [DOI] [PubMed] [Google Scholar]
- 67.Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol. 2008;14:3937–3947. doi: 10.3748/wjg.14.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 69.Bingham SA. Diet and colorectal cancer prevention. Biochem Soc Trans. 2000;28:12–16. doi: 10.1042/bst0280012. [DOI] [PubMed] [Google Scholar]
- 70.Hope ME, Hold GL, Kain R, El-Omar EM. Sporadic colorectal cancer--role of the commensal microbiota. FEMS Microbiol Lett. 2005;244:1–7. doi: 10.1016/j.femsle.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 71.Candela M, Guidotti M, Fabbri A, Brigidi P, Franceschi C, Fiorentini C. Human intestinal microbiota: cross-talk with the host and its potential role in colorectal cancer. Crit Rev Microbiol. 2011;37:1–14. doi: 10.3109/1040841X.2010.501760. [DOI] [PubMed] [Google Scholar]
- 72.Plottel CS, Blaser MJ. Microbiome and malignancy. Cell Host Microbe. 2011;10:324–335. doi: 10.1016/j.chom.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012;10:575–582. doi: 10.1038/nrmicro2819. [DOI] [PubMed] [Google Scholar]
- 74.Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, Lam V, Potentier MS, Ng K, Bawa M, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis. 2011;17:1359–1372. doi: 10.1002/ibd.21478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Horst D, Reu S, Kriegl L, Engel J, Kirchner T, Jung A. The intratumoral distribution of nuclear beta-catenin is a prognostic marker in colon cancer. Cancer. 2009;115:2063–2070. doi: 10.1002/cncr.24254. [DOI] [PubMed] [Google Scholar]
- 76.Mai V, McCrary QM, Sinha R, Glei M. Associations between dietary habits and body mass index with gut microbiota composition and fecal water genotoxicity: an observational study in African American and Caucasian American volunteers. Nutr J. 2009;8:49. doi: 10.1186/1475-2891-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McConnell BB, Yang VW. The Role of Inflammation in the Pathogenesis of Colorectal Cancer. Curr Colorectal Cancer Rep. 2009;5:69–74. doi: 10.1007/s11888-009-0011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marchesi JR, Dutilh BE, Hall N, Peters WH, Roelofs R, Boleij A, Tjalsma H. Towards the human colorectal cancer microbiome. PLoS One. 2011;6:e20447. doi: 10.1371/journal.pone.0020447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7:e39743. doi: 10.1371/journal.pone.0039743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu N, Yang X, Zhang R, Li J, Xiao X, Hu Y, Chen Y, Yang F, Lu N, Wang Z, et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb Ecol. 2013;66:462–470. doi: 10.1007/s00248-013-0245-9. [DOI] [PubMed] [Google Scholar]
- 81.Wang X, Huycke MM. Extracellular superoxide production by Enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology. 2007;132:551–561. doi: 10.1053/j.gastro.2006.11.040. [DOI] [PubMed] [Google Scholar]
- 82.Zheng J, Meng J, Zhao S, Singh R, Song W. Campylobacter-induced interleukin-8 secretion in polarized human intestinal epithelial cells requires Campylobacter-secreted cytolethal distending toxin- and Toll-like receptor-mediated activation of NF-kappaB. Infect Immun. 2008;76:4498–4508. doi: 10.1128/IAI.01317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–215. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McCoy AN, Araújo-Pérez F, Azcárate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium is associated with colorectal adenomas. PLoS One. 2013;8:e53653. doi: 10.1371/journal.pone.0053653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martínez I, Perdicaro DJ, Brown AW, Hammons S, Carden TJ, Carr TP, Eskridge KM, Walter J. Diet-induced alterations of host cholesterol metabolism are likely to affect the gut microbiota composition in hamsters. Appl Environ Microbiol. 2013;79:516–524. doi: 10.1128/AEM.03046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294:1–8. doi: 10.1111/j.1574-6968.2009.01514.x. [DOI] [PubMed] [Google Scholar]
- 89.Fanning S, Hall LJ, Cronin M, Zomer A, MacSharry J, Goulding D, Motherway MO, Shanahan F, Nally K, Dougan G, et al. Bifidobacterial surface-exopolysaccharide facilitates commensal-host interaction through immune modulation and pathogen protection. Proc Natl Acad Sci USA. 2012;109:2108–2113. doi: 10.1073/pnas.1115621109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Claesson MJ, Jeffery IB, Conde S, Power SE, O’Connor EM, Cusack S, Harris HM, Coakley M, Lakshminarayanan B, O’Sullivan O, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488:178–184. doi: 10.1038/nature11319. [DOI] [PubMed] [Google Scholar]
- 91.Bliska JB, van der Velden AW. Salmonella “sops” up a preferred electron receptor in the inflamed intestine. MBio. 2012;3:e00226–e00212. doi: 10.1128/mBio.00226-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huycke MM, Abrams V, Moore DR. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis. 2002;23:529–536. doi: 10.1093/carcin/23.3.529. [DOI] [PubMed] [Google Scholar]
- 93.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Toprak NU, Yagci A, Gulluoglu BM, Akin ML, Demirkalem P, Celenk T, Soyletir G. A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin Microbiol Infect. 2006;12:782–786. doi: 10.1111/j.1469-0691.2006.01494.x. [DOI] [PubMed] [Google Scholar]
- 95.Ahmed S, Macfarlane GT, Fite A, McBain AJ, Gilbert P, Macfarlane S. Mucosa-associated bacterial diversity in relation to human terminal ileum and colonic biopsy samples. Appl Environ Microbiol. 2007;73:7435–7442. doi: 10.1128/AEM.01143-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shen XJ, Rawls JF, Randall T, Burcal L, Mpande CN, Jenkins N, Jovov B, Abdo Z, Sandler RS, Keku TO. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010;1:138–147. doi: 10.4161/gmic.1.3.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sears CL, Pardoll DM. Perspective: alpha-bugs, their microbial partners, and the link to colon cancer. J Infect Dis. 2011;203:306–311. doi: 10.1093/jinfdis/jiq061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Boleij A, Tjalsma H. The itinerary of Streptococcus gallolyticus infection in patients with colonic malignant disease. Lancet Infect Dis. 2013;13:719–724. doi: 10.1016/S1473-3099(13)70107-5. [DOI] [PubMed] [Google Scholar]
- 99.Wentling GK, Metzger PP, Dozois EJ, Chua HK, Krishna M. Unusual bacterial infections and colorectal carcinoma--Streptococcus bovis and Clostridium septicum: report of three cases. Dis Colon Rectum. 2006;49:1223–1227. doi: 10.1007/s10350-006-0576-4. [DOI] [PubMed] [Google Scholar]
- 100.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 101.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 102.Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Göktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 103.Femia AP, Luceri C, Toti S, Giannini A, Dolara P, Caderni G. Gene expression profile and genomic alterations in colonic tumours induced by 1,2-dimethylhydrazine (DMH) in rats. BMC Cancer. 2010;10:194. doi: 10.1186/1471-2407-10-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kraus S, Arber N. Inflammation and colorectal cancer. Curr Opin Pharmacol. 2009;9:405–410. doi: 10.1016/j.coph.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 105.Lee SH, Hu LL, Gonzalez-Navajas J, Seo GS, Shen C, Brick J, Herdman S, Varki N, Corr M, Lee J, et al. ERK activation drives intestinal tumorigenesis in Apc(min/+) mice. Nat Med. 2010;16:665–670. doi: 10.1038/nm.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rutter M, Saunders B, Wilkinson K, Rumbles S, Schofield G, Kamm M, Williams C, Price A, Talbot I, Forbes A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology. 2004;126:451–459. doi: 10.1053/j.gastro.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 107.Gupta RB, Harpaz N, Itzkowitz S, Hossain S, Matula S, Kornbluth A, Bodian C, Ullman T. Histologic inflammation is a risk factor for progression to colorectal neoplasia in ulcerative colitis: a cohort study. Gastroenterology. 2007;133:1099–105; quiz 1340-1. doi: 10.1053/j.gastro.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stecher B, Hardt WD. The role of microbiota in infectious disease. Trends Microbiol. 2008;16:107–114. doi: 10.1016/j.tim.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 109.Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:119–129. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 110.Uronis JM, Mühlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One. 2009;4:e6026. doi: 10.1371/journal.pone.0006026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schwabe RF, Wang TC. Cancer. Bacteria deliver a genotoxic hit. Science. 2012;338:52–53. doi: 10.1126/science.1229905. [DOI] [PubMed] [Google Scholar]
- 112.Lax AJ. Opinion: Bacterial toxins and cancer--a case to answer? Nat Rev Microbiol. 2005;3:343–349. doi: 10.1038/nrmicro1130. [DOI] [PubMed] [Google Scholar]
- 113.Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu S, Morin PJ, Maouyo D, Sears CL. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology. 2003;124:392–400. doi: 10.1053/gast.2003.50047. [DOI] [PubMed] [Google Scholar]
- 115.Sears CL. Enterotoxigenic Bacteroides fragilis: a rogue among symbiotes. Clin Microbiol Rev. 2009;22:349–369. doi: 10.1128/CMR.00053-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Housseau F, Sears CL. Enterotoxigenic Bacteroides fragilis (ETBF)-mediated colitis in Min (Apc+/-) mice: a human commensal-based murine model of colon carcinogenesis. Cell Cycle. 2010;9:3–5. doi: 10.4161/cc.9.1.10352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lara-Tejero M, Galán JE. Cytolethal distending toxin: limited damage as a strategy to modulate cellular functions. Trends Microbiol. 2002;10:147–152. doi: 10.1016/s0966-842x(02)02316-8. [DOI] [PubMed] [Google Scholar]
- 118.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 119.Ge Z, Rogers AB, Feng Y, Lee A, Xu S, Taylor NS, Fox JG. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell Microbiol. 2007;9:2070–2080. doi: 10.1111/j.1462-5822.2007.00939.x. [DOI] [PubMed] [Google Scholar]
- 120.Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, Pezet D, Bonnet R. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One. 2013;8:e56964. doi: 10.1371/journal.pone.0056964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 122.Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A. 2010;107:11537–11542. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fiorentini C, Fabbri A, Flatau G, Donelli G, Matarrese P, Lemichez E, Falzano L, Boquet P. Escherichia coli cytotoxic necrotizing factor 1 (CNF1), a toxin that activates the Rho GTPase. J Biol Chem. 1997;272:19532–19537. doi: 10.1074/jbc.272.31.19532. [DOI] [PubMed] [Google Scholar]
- 124.Falzano L, Filippini P, Travaglione S, Miraglia AG, Fabbri A, Fiorentini C. Escherichia coli cytotoxic necrotizing factor 1 blocks cell cycle G2/M transition in uroepithelial cells. Infect Immun. 2006;74:3765–3772. doi: 10.1128/IAI.01413-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fabbri A, Travaglione S, Ballan G, Loizzo S, Fiorentini C. The cytotoxic necrotizing factor 1 from E. coli: a janus toxin playing with cancer regulators. Toxins (Basel) 2013;5:1462–1474. doi: 10.3390/toxins5081462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Boyer L, Magoc L, Dejardin S, Cappillino M, Paquette N, Hinault C, Charriere GM, Ip WK, Fracchia S, Hennessy E, et al. Pathogen-derived effectors trigger protective immunity via activation of the Rac2 enzyme and the IMD or Rip kinase signaling pathway. Immunity. 2011;35:536–549. doi: 10.1016/j.immuni.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Holmes E, Li JV, Athanasiou T, Ashrafian H, Nicholson JK. Understanding the role of gut microbiome-host metabolic signal disruption in health and disease. Trends Microbiol. 2011;19:349–359. doi: 10.1016/j.tim.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 128.Degirolamo C, Modica S, Palasciano G, Moschetta A. Bile acids and colon cancer: Solving the puzzle with nuclear receptors. Trends Mol Med. 2011;17:564–572. doi: 10.1016/j.molmed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 129.Carbonero F, Benefiel AC, Gaskins HR. Contributions of the microbial hydrogen economy to colonic homeostasis. Nat Rev Gastroenterol Hepatol. 2012;9:504–518. doi: 10.1038/nrgastro.2012.85. [DOI] [PubMed] [Google Scholar]
- 130.Windey K, De Preter V, Louat T, Schuit F, Herman J, Vansant G, Verbeke K. Modulation of protein fermentation does not affect fecal water toxicity: a randomized cross-over study in healthy subjects. PLoS One. 2012;7:e52387. doi: 10.1371/journal.pone.0052387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Seitz HK, Stickel F. Acetaldehyde as an underestimated risk factor for cancer development: role of genetics in ethanol metabolism. Genes Nutr. 2010;5:121–128. doi: 10.1007/s12263-009-0154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Leonel AJ, Alvarez-Leite JI. Butyrate: implications for intestinal function. Curr Opin Clin Nutr Metab Care. 2012;15:474–479. doi: 10.1097/MCO.0b013e32835665fa. [DOI] [PubMed] [Google Scholar]
- 133.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, Glimcher LH. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453:620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 136.Sansonetti PJ, Di Santo JP. Debugging how bacteria manipulate the immune response. Immunity. 2007;26:149–161. doi: 10.1016/j.immuni.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 137.Kamada N, Seo SU, Chen GY, Núñez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13:321–335. doi: 10.1038/nri3430. [DOI] [PubMed] [Google Scholar]
- 138.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Biagi E, Candela M, Turroni S, Garagnani P, Franceschi C, Brigidi P. Ageing and gut microbes: perspectives for health maintenance and longevity. Pharmacol Res. 2013;69:11–20. doi: 10.1016/j.phrs.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 141.Boyle P, Ferlay J. Mortality and survival in breast and colorectal cancer. Nat Clin Pract Oncol. 2005;2:424–425. doi: 10.1038/ncponc0288. [DOI] [PubMed] [Google Scholar]
- 142.Venturi V, da Silva DP. Incoming pathogens team up with harmless ‘resident’ bacteria. Trends Microbiol. 2012;20:160–164. doi: 10.1016/j.tim.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 143.Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, Núñez G. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012;336:1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Marks LR, Davidson BA, Knight PR, Hakansson AP. Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. MBio. 2013;4 doi: 10.1128/mBio.00438-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang C, Li S, Yang L, Huang P, Li W, Wang S, Zhao G, Zhang M, Pang X, Yan Z, et al. Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat Commun. 2013;4:2163. doi: 10.1038/ncomms3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ishikawa H, Akedo I, Otani T, Suzuki T, Nakamura T, Takeyama I, Ishiguro S, Miyaoka E, Sobue T, Kakizoe T. Randomized trial of dietary fiber and Lactobacillus casei administration for prevention of colorectal tumors. Int J Cancer. 2005;116:762–767. doi: 10.1002/ijc.21115. [DOI] [PubMed] [Google Scholar]
- 147.Pool-Zobel BL. Inulin-type fructans and reduction in colon cancer risk: review of experimental and human data. Br J Nutr. 2005;93 Suppl 1:S73–S90. doi: 10.1079/bjn20041349. [DOI] [PubMed] [Google Scholar]
- 148.Rafter J, Bennett M, Caderni G, Clune Y, Hughes R, Karlsson PC, Klinder A, O’Riordan M, O’Sullivan GC, Pool-Zobel B, et al. Dietary synbiotics reduce cancer risk factors in polypectomized and colon cancer patients. Am J Clin Nutr. 2007;85:488–496. doi: 10.1093/ajcn/85.2.488. [DOI] [PubMed] [Google Scholar]
- 149.Davis CD, Milner JA. Gastrointestinal microflora, food components and colon cancer prevention. J Nutr Biochem. 2009;20:743–752. doi: 10.1016/j.jnutbio.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ventura M, Turroni F, Motherway MO, MacSharry J, van Sinderen D. Host-microbe interactions that facilitate gut colonization by commensal bifidobacteria. Trends Microbiol. 2012;20:467–476. doi: 10.1016/j.tim.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 151.Candela M, Perna F, Carnevali P, Vitali B, Ciati R, Gionchetti P, Rizzello F, Campieri M, Brigidi P. Interaction of probiotic Lactobacillus and Bifidobacterium strains with human intestinal epithelial cells: adhesion properties, competition against enteropathogens and modulation of IL-8 production. Int J Food Microbiol. 2008;125:286–292. doi: 10.1016/j.ijfoodmicro.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 152.Ruas-Madiedo P, Medrano M, Salazar N, De Los Reyes-Gavilán CG, Pérez PF, Abraham AG. Exopolysaccharides produced by Lactobacillus and Bifidobacterium strains abrogate in vitro the cytotoxic effect of bacterial toxins on eukaryotic cells. J Appl Microbiol. 2010;109:2079–2086. doi: 10.1111/j.1365-2672.2010.04839.x. [DOI] [PubMed] [Google Scholar]
- 153.Carey CM, Kostrzynska M, Ojha S, Thompson S. The effect of probiotics and organic acids on Shiga-toxin 2 gene expression in enterohemorrhagic Escherichia coli O157: H7. J Microbiol Methods. 2008;73:125–132. doi: 10.1016/j.mimet.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 154.Bayoumi MA, Griffiths MW. In vitro inhibition of expression of virulence genes responsible for colonization and systemic spread of enteric pathogens using Bifidobacterium bifidum secreted molecules. Int J Food Microbiol. 2012;156:255–263. doi: 10.1016/j.ijfoodmicro.2012.03.034. [DOI] [PubMed] [Google Scholar]