ISSN: 1837-9664Journal of Cancer

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(8):1394-1402. doi:10.7150/jca.23835 This issue Cite
Research Paper
Protein arginine methyltransferase 8 gene enhances the colon cancer stem cell (CSC) function by upregulating the pluripotency transcription factor
Haishan Lin1#, Bin Wang2#, Jing Yu1, Jing Wang1, Qin Li1, Bangwei Cao1 ![]()
1. Department of Oncology, Beijing Friendship Hospital, Capital Medical University, Beijing 100050, China
2. Department of Medical Administration, Beijing Friendship Hospital, Capital Medical University, Beijing 100050, China
#There authors contributed equally to this work and should be considered co-first authors.
Citation:
Lin H, Wang B, Yu J, Wang J, Li Q, Cao B. Protein arginine methyltransferase 8 gene enhances the colon cancer stem cell (CSC) function by upregulating the pluripotency transcription factor. J Cancer 2018; 9(8):1394-1402. doi:10.7150/jca.23835. https://www.jcancer.org/v09p1394.htm
Other stylesAbstract
Objective: Cancer stem cells play a crucial role in tumor multidrug resistance and metastasis, which can produce heterogeneous tumor cells and have self-renewal ability. The related literature reported that PRMT8 was overexpressed in tumor stem cells and pluripotent stem cells. However, it's unclear how PRMT8 acts on the stemness of colon tumor cells. This study is designed to detect functions by transfecting with PRMT8 plasmid to colon cancer cells.
Methods: In this study we investigated colon cancer cell sphere and its differential expression of PRMT8 compared with colon cancer cells grown by static adherence. RKO Sphere formation assay was used to identify CSCs and verified PRMT8 and pluripotent transcription factors SOX2, OCT4, Nanog expression level in colon cell sphere. Colon cancer cell HCT-8 and RKO up-regulated PRMT8 expression by being transfected with PRMT8 plasmid to evaluate its effect on the stemness of colon tumor cell.
Results: In RKO cell sphere, stem cell surface marker CD133 and CD44 were highly expressed. And PRMT8, SOX2, OCT4 and Nanog were also highly expressed in RKO cell sphere. After PRMT8 was up-regulated in HCT-8 and RKO cells, flow cytometry proved that PRMT8 group cells have a significant increase of the side population (SP) cells with cancer stem cell surface markers CD133 and CD44. And overexpression of PRMT8 in HCT-8 and RKO cells facilitated their aggressive traits, which contained proliferation, invasion and migration, as well as leading to their drug resistance. PRMT8 may play a role in colon cancer stem cells (CSC) through its regulation of pluripotent transcription factors, such as Nanog Homeobox (Nanog), octamer-binding transcription factor-4 (Oct4) and SRY-related high-mobility-group(HMG)-box protein-2 (Sox2).
Conclusion: PRMT8 may promote the formation of colon cancer stem cells and, thus, be considered a potential therapeutic target for the treatment of malignant colon tumor.
Keywords: Protein arginine methyltransferase 8, Colon cancer stem cell, Pluripotency transcription factor
Introduction
Colon cancer is the fourth highest fatal malignant tumor reported by world health organization (WHO) [1]. With the development of tumor therapy, some promising emerging therapies such as targeted therapy and immunotherapy are active in first-line treatment. However, the treatment for colon cancer has not progressed that much, there is a few targeted or immunotherapy drugs that can be included in the first-line treatment for colon cancer, even the types of traditional chemical remedies available are limited. Therefore, it is urgent to find the breakout of colon cancer treatment.
The tumor stem cell hypothesis was first proposed by Mackillop in 1983, and he believes that there may be a small subset of cells in all tumors that have the special function of stem cells [2]. The theory has been gradually verified in the course of continuous research on tumor research. The American Association for Cancer Research (AACR) defines tumor stem cells as cells that are self-renewing and can produce heterogenous tumor cells [3]. Cancer stem cell (CSC) has characteristics of self-renewal, high oncogenicity, differentiation potential and drug resistance. Tumor stem cell self-renewal maintained sustained growth and accumulated in tumor gene mutations. It is these genetic mutations that can lead to the excessive proliferation of tumor cells, and even spread [4]. Human colon CSCs are primarily characterized by cluster of differentiation (CD)44+/CD133+/high cells, sphere formation and active aldehyde dehydrogenase [5]. Another feature of CSCs is overexpression of stem cell-associated genes, including octamer-binding transcription factor 4 (Oct-4) and sex determining region Y-box 2 (Sox-2) [6].
Protein Arginine Methyltransferases (PRMT) family is a group of enzymes which can catalyze to add methyl groups to arginine residues. It consists of nine members, classified as type I, type II or type III according to their capacity of catalyzing the formation of ADMA, SDMA or MMA, respectively. PRMT1, 2, 3, 4, 6 and 8 belong to type I, whereas PRMT5 and 9 belong to type II. PRMT1 is responsible for at least 85% of all arginine methylation reactions in the cell and is ubiquitously expressed in mammalian cells [7]. Regulation and cellular substrates of PRMT8 are poorly understood, some research show that PRMT8 is expressed in mouse ESC and iPSC [8,9]. In this study, related tests were used to evaluate the regulation effect of PRMT8 on the stemness of colon cancer cells.
Materials and Methods
Cell culture
Human colon cancer cell lines, RKO and HCT-8, were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). RKO and HCT-8 cells were cultured in DMEM supplemented with 10% fetal bovine serum (Corning, USA) and 1% penicillin/streptomycin solution (Invitrogen). Both cell lines were cultured at 37˚C under a humidified atmosphere of 5% CO2.
Sphere Formation Assay
RKO cells were plated at a density of 2,000 cells/well on an ultralow attachment 6-well plate (Corning Incorporated, Corning, NY, USA.) in serum-free DMEM medium (Corning, USA.) supplemented with 20 ng/ml of Recombinant Human FGF-basic (b FGF) (PeproTech, USA), 20 ng/ml of epidermal growth factor(EGF) (PeproTech USA) and 50X B-27 (PeproTech, USA). The number of spheres was counted after a 7-day incubation (at 37°C in atmosphere containing 5% CO2). The culture medium was changed fresh in half of the amount every 3 days.
Immunofluorescence
Immunofluorescence detection was used to test CD133 and CD44 on RKO cell spheres. CD133 and CD44 were CSC surface marker. After RKO cell sphere formation, the sphere was seed to common attachment 6-well plate in medium containing 10% calf bovine serum. When RKO cell sphere adherence, the cells were rinsed with PBS and then fixed in 4% paraformaldehyde before permeabilization with 0.3% Triton X-100. After blocking with 5% BSA (Sigma), cells were incubated with diluted primary antibody for PE flurochrome-CD133 and FITC flurochrome-CD44 for 2 hours at room temperature (BD Bioscience, San Diego, USA). Finally, the samples were mounted in mounting medium containing DAPI (ThermoFisher Scientific). Three random fields were examined at a magnification of ×63 by a Leica TCS SP5 Confocal Laser Scanning Microscope.
Transfection
Cells were seeded in 6-well plates 1 day before transfection with cell concentration about 50,000/well of RKO cell and 45,000/well of HCT-8 cell. When cells confluence reached 70%-80%, transfection was conducted using LipofectamineTM2000 (Invitrogen). HCT-8 and RKO cells were transfected with PRMT8-pMCV (Origene, USA) or control vector-pMCV (Origene, USA). During the process of transfection, cells were cultured in serum-free DMEM. The medium was replaced 6 hours after transfection and the cells were incubated in a cell incubator.
The Side Population (SP) Cells Assay
For phenotypic characterization, isolated cells were stained with monoclonal antibodies tagged with PE flurochrome -CD133(BD Bioscience, San Diego, USA) and tagged with APC fulrochrome - CD44(BD Bioscience, San Diego, USA) and incubated at 4°C for 45 min. Cells were acquired on FACS Canto II (BD Bioscience, San Diego, USA). Data were analyzed using FACS Diva™ software. (BD Bioscience). For each sample, data of at least 10.000 events in the gate were recorded.
RNA Extraction and Quantitative-polymerase Chain Reaction (q-PCR)
Total RNA was extracted from cultured cells using TRI reagent (Sigma-Aldrich, Irvine, UK) according to the manufacturer's instructions and quantified using a Nanodrop spectrophotometer. cDNA samples were generated from the total RNA using GoScript™ Reverse Transcription System kit (Promega, Madison, WI, USA). Subsequently, q-PCR was conducted using a FAST SYBR™ Green Master Mix (Thermo Fisher Scientific) on an ABI Prism 7700 Sequence Detector (Applied Biosystems Inc., Foster City, CA, USA). The following primers were used: Nanog: forward, 5'-TgAACATgCAACCTgAAgACgT-3' and reverse, 5'-TgATTAggCTCCAACCATACTCC-3'. Oct4: forward, 5'-TATgCAAAgCAgAAACCCTCgTgC-3' and reverse, 5'-TTCgggCACTgCAggAACAAATTC-3'. SOX2: forward, 5'-gCCgAgTggAAACTTTTgTCg-3' and reverse, 5'-ggCAgCgTgTACTTATCCTTCT-3'. PRMT8: forward, 5'-GGAGCCTCTAGTGGACATCG-3' and reverse, 5'-CCCATTTTCTTGTGGCACTT-3'. GAPDH: forward, 5'-GGCTGCTTTTAACTCTGGTA-3' and reverse, 5'-ACTGTGGTCATGAGTCCTT-3'. The following cycling conditions were used: 25˚C for 10min, 37˚C for 120s, followed by 85˚C for 5 min. 2(-ΔΔCt) was used to data statistics of qPCR.
Western Blotting Analysis
Whole cell protein was extracted and quantified by NanoDrop 2000c spectrophotometer (Thermo Scientific, Waltham, MA, USA) using the BCA protein assay kit. After separation by SDSPAGE (a 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis), the proteins were then transferred to a PVDF membrane (Millipore). Proteins were probed with primary antibodies anti-PRMT8, anti-SOX2, anti-OCT4 or anti-Nanog (1:1000; Abcam) and incubated with the corresponding horseradish peroxidase-conjugated secondary antibodies (1:2000; Santa Cruz Biotechnology). The proteins were detected with Z-ECL chemiluminescence kit (Luminata Forte; Millipore, Hertfordshire, UK). GAPDH was used as an internal control. The intensity values were analyzed using the Image J software (NIH).
Cell Proliferation Assay
RKO and HCT-8 cell proliferation were estimated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (Promega Corporation). The proliferation of RKO and HCT-8 cells 24 hours after being transfected with the above mentioned PRMT8-pCMV and null plasmids in 96-well plates with 5000 cells per pore were monitored every day for 4 days. At the indicated time points of these four days, after adding into the wells with 50 µL MTT working solution, the 96-well plate was incubated at 37°C for 4 hours. Then, the culture medium and MTT were removed and the formazan crystals formed by MTT treatment solubilized in 150 µL dimethylsulfoxide (DMSO) solution for 20 minutes. Absorbance at 570 nm was then measured using a microplate reader (Bio-Rad Laboratories Inc., Hercules, CA, USA). Each treatment group was composed of 6 separate wells.
Colony Formation Assay
After 48 hours, RKO and HCT-8 cells transfected with PRMT8-pCMV and null plasmid were mixed with DMEM containing 10% FBS, plated in six-well culture dishes at a density of 1,000 cells/well, and coated with 3ml DMEM containing 10% FBS, in triplicate. The culture medium was replaced with fresh DMEM containing 10% FBS every 3 days and after 14 days cultured at 37°C, under a humidified atmosphere of 5% CO2. The resulting colonies were fixed with methanol for 5 minutes, and then stained with 0.005% crystal violet (Sigma-Aldrich Co., St Louis, MO, USA). The number of clearly visible colonies (diameter >50 µm) in three random microscopic fields was counted, and the images were captured under a microscope (Leica Microsystems, Wetzlar, Germany) and analyzed using ImageJ software.
In Vitro Wound Healing Assay
Approximately 40,000 cells were seeded into a 24-well plate and allowed to reach confluency. The cell monolayer was scraped using a P10 pipette tip. Photographs were taken at 0 and 6 and 12 hours after wounding. Migration distances were measured using the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
In Vitro Transwell Invasion and Migration Assay
A Transwell insert with 8 μm pores was coated with 50 μg of Matrigel for the invasion assay. Forty thousand cells were seeded into each insert for migration and invasion assay, respectively. After 48 hours of incubation, DIPI (HermoFisher Scientific) was added and the transwell inserts were incubated further for 1 h at 37˚C. Fluorescence was determined at a wavelength of ex490/em510 nm using the GloMax®-Multi Detection System (Promega).
Cell Viability Assay for Chemoresistance
Cells were seeded in 96-well plates and then treated with the indicated concentrations of cisplatin on the second day. After this, the cells were cultured for another 36 hours and then subjected to MTT assay. The live cell number was estimated as 5000 cells per pore. For the MTT assay, 20μl of MTT (5 mg/ml) was added to each sample followed by incubation at 37˚C for 4 hours. Following this, the solution was iscarded, and 100μl of dimethyl sulfoxide was added. Cell viability was determined at 495nm.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism software (version 5.0). Data are presented as the mean ± standard deviation. A student's test was used when 2 groups were being compared. P<0.05 was considered to indicate a statistically significant difference.
Results
RKO Spheres Exhibit Characteristics of CSCs
RKO spheres derived from parental cells, under anchorage-independent conditions and in the absence of FBS, exhibited characteristics of CSCs (Fig. 1A). Immunofluorescence showed spheres common expressed CD133and CD44 on their surface (Fig. 1B). q-PCR show the PRMT8 and pluripotent transcription factors Oct4 and Sox2 and Nanog mRNA were up-regulated. These results indicate that the spheres derived from RKO cells are CSCs (P<0.05; Fig. 1C). In addition, western blot analysis demonstrated that stem cell marker Oct-4, Sox2, Nanog and PRMT8 were overexpressed in spheres when compared with their parental cells (Fig. 1D).
Figure 1

Human cancer stem cells were identified using a sphere formation assay and immunofluorescence, and their expression of pluripotent transcription factors mRNA and proteins. (A) Representative morphology of spheres derived from the human colon cancer cell RKO. The four images represent the change of morphology of spheres after sphere formation 0h 12h 24h 36h. (B) Immunofluorescence analysis of the expression of CD133 and CD44 in spheres were common. Results are demonstrated as representative images. The blue dot indicates DIPI, the red dot indicates CD133, the green dot indicates CD44. (C) mRNA levels of Sox2, Oct4, Nanog and PRMT8 in spheres were much higher than their parental cells. (*P<0.05, **P<0.01, ***P<0.001) (D) Protein levels of Sox2, Oct-4, Nanog, PRMT8 in spheres and their parental cells, measured by western blot analysis. These proteins were overexpressed in spheres compared with their parental cells.
Overexpression of PRMT8 in HCT-8 and RKO Cell Lines and Its Regulatory Genes
PRMT8 gene expression was overexpressed by transfecting PRMT8-pCMV. The overexpression of PRMT8 in HCT-8 and RKO cells was verified using qPCR compared to their corresponding vector control cells. And Oct4 and Nanog mRNA level were up-regulated after RKO and HCT-8 PRMT8 overexpression. Sox2 mRNA seem to have no chance when RKO and HCT-8 cell PRMT8 overexpression (Figure 2A and 2B). The level of PRMT8 protein also confirmed by western blog that there was up-regulation in HCT-8 and RKO cells. Pluripotent transcription factors Sox2, Nanog and Oct4 protein level all increased after colon cells PRMT8 overexpression (Figure 2C).
Overexpression of PRMT8 up-regulated CSC Phenotype Cells
PRMT8 overexpression in RKO exhibited an increased level of the set of cell with a CD44+/CD133+/high phenotype (Figure 3).
Overexpression of PRMT8 Accelerated Cell Proliferation and Colony Formation in Colon cancer
Increased PRMT8 expression led to significant promotion of cell proliferation in HCT-8 and RKO (p<0.05, Figure 4A). To elucidate whether PRMT8 can regulate HCT-8 cell growth, soft agar colony formation assays were carried out. Cells transfected with PRMT8-pCMV showed increased colony formation rate compared with those transfected with the vector only control (Figure 4B).
PRMT8 Promoted Colon Cancer Cell Invasion and Migration
The migration capacity of the HCT-8 cells use the scratch wound-healing assay. The overexpression of PRMT8 in HCT-8 cells increased the rate of the wound healing from 5.78% to 10.34% 12 hours after scratch wounding. Using the ECIS system, we were able to quantify the attachment and post-wound migration of cells in a high throughput real-time manner (Figure 5A and 5B). Cell invasion was measured by Transwell assays in vitro and found that augmented PRMT8 expression greatly improved the migratory and invasive behaviors of HCT-8 and RKO cells (Figure 5C and D).
Figure 2

Identification of pluripotent transcription factors expression after PRMT8 overexpression in colon cancer cells (A) qPCR showing RKO cell after PRMT8 overexpression showed a significantly increase in the expression level of pluripotent transcription factors Oct4, Nanog mRNA level, while, Sox2 had no changes of the control cells. (*P<0.05, **P<0.01, ***P<0.001) (B) Oct4, Nanog mRNA also increased in HCT-8 cell after PRMT8 overexpression. Sox2 mRNA level expression had no difference in HCT-8 after PRMT8 overexpression. (*P<0.05, **P<0.01, ***P<0.001) (C) Western blotting shows the up-regulation of pluripotent transcription factors protein Oct4, Nanog, Sox2 after PRMT8 overexpression in RKO and HCT-8.
Figure 3

Cell side population analysis of HCT-8 cells after PRMT8 overexpression (A) Flow cytometry shows the percentage of cell expressed CD133 and CD44 increased significantly after HCT-8 overexpression PRMT8. (B) The percentage of CD44+ and CD133+ cells following transfection with PRMT8-pCMV was increased. Results are demonstrated as representative images of three independent experiments. (*P<0.05, **P<0.01, ***P<0.001)
Cell viability Assay for Chemoresistance
The MTT cell viability assay was used to study the cytotoxic effect of pirarubicin and cisplatin in the two cancer cell lines RKO and HCT-8. Overexpression of PRMT8 exhibited a dose dependent cytotoxicity on the cancer cell lines. Selective cytotoxicity of cisplatin to HCT-8 and RKO cells as evidenced by the high IC50 values 11.24 μg mL-1 and 6.53 μg mL-1 on the control cancer cell line. Significant chemoresistance was observed on PRMT8 overexpression colon cancer cells with IC50 values of 6.74 μg mL-1 and 4.18 μg mL-1 (Figure 5).
Conclusion
Protein methylation modification involved in a variety of disease processes [10-15]. The PRMT family is the family of protein-arginine methyltransferase, which found to have an especial role of pluripotent stem cell [16-20]. Sox2, one of pluripotent transcription factors, can modulate PRMT8 promote activity, due to binding to the stem consensus sites [21]. PRMT1 can make a methylation modification on Axin at R378 to negatively regulate WNT pathway [22]. Moreover, WNT pathway seem to confer resistance and survival to CSCs in human cancer [17] and PRMT8 is an important paralog of this gene PRMT1[23]. Therefore, we can speculate that PRMT8 may play a role in tumor stem cells. So far, the researches about PRMT8 mainly focus on pluripotent or unipotent stem cell rather than cancer stem cell. This study is a preliminary research about PRMT8 effecting on colon cancer stem cell via a relatively new pathway.
Figure 4
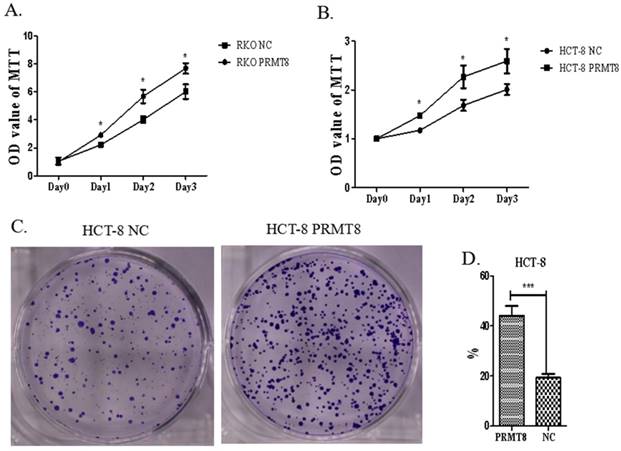
Evaluation of proliferation of colon cancer cells after PRMT8 overexpression. (A) MTT used to proliferation assay showed RKO cell after PRMT8 overexpression increase in cell proliferation. (*P<0.05, **P<0.01, ***P<0.001) (B) HCT-8 cell after PRMT8 overexpression show an increasing in cell proliferation tested by MTT. (*P<0.05, **P<0.01, ***P<0.001) (C) Colony formation teat show HCT-8 after PRMT8 overexpression increased in colony formation rate. (*p<0.05. **p<0.01. ***p<0.001) (D) Statistical results showed that colony formation rate of HCT-8 after PRMT8 overexpression was significantly higher than that in the control group. (*p<0.05. **p<0.01. ***p<0.001)
Figure 5
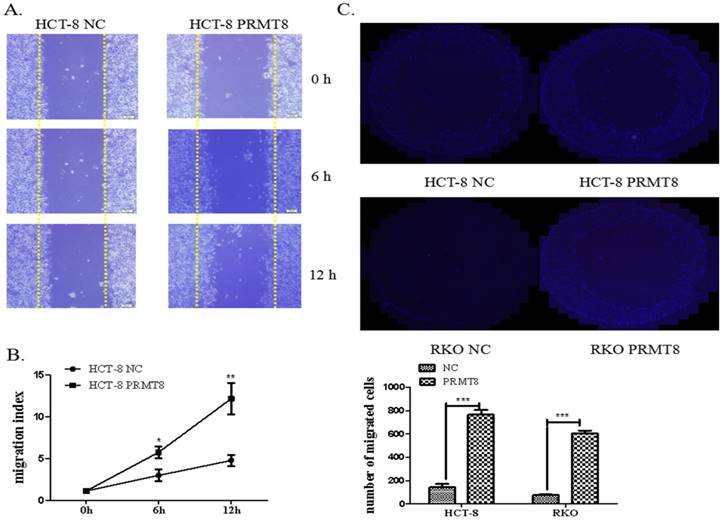
Evaluation of migration and invasion of colon cancer cells after PRMT8 overexpression (A) Scratch wound healing showing the promotion of the migration capacity in the HCT-8 cells with PRMT8 overexpression. Migration index (%) = [(the initialized width of the scratch) - (the final width of the scratch)] / (the initialized width of the scratch. (*P<0.05, **P<0.01, ***P<0.001) (B) Transwell shows HCT-8 and RKO after PRMT8 overexpression promote invasion capital significantly. After 48h migration incubation, HCT-8 and RKO cell with PRMT8 overexpression show more cells have traversed the tiny holes of Transwell plate; Counts of cells under a microscope compared with the control group. (*p < 0.05. **p < 0.01. ***p < 0.001)
Figure 6
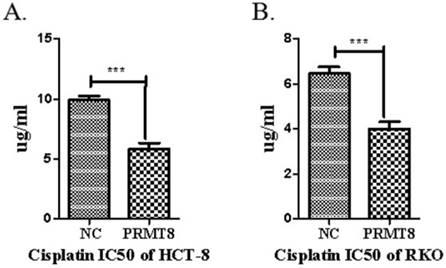
Chemoresistance analyze RKO and HCT-8 cell after PRMT8 overexpression. The 48 h IC50 of DDP in HCT-8 and RKO control cell and HCT-8 and RKO after PRMT8 overexpression. Results are presented as the mean ± standard deviation (n=3). (*p < 0.05. **p < 0.01. ***p < 0.001)
In this research, PRMT8 gene was overexpressed through transfecting PRMT8-pCMV to colon cancer cells. The expression quantity of PRMT8 gene was detected through qPCR and WB. The high expression of PRMT8 gene in colon cancer cells enhanced more characteristics of tumor stem cells. The all results of this research suggested PRMT8 gene may induce colon cancer cells expressing stem cell phenotype, with the high tumorigenic ability, drug resistance and the improvement of tumor cell migration and invasion capacity. PRMT8 was confirmed high expression in colon cancer stem cell. According to the qPCR and WB results of this research, PRMT8 gene overexpression can lead to up-expression of pluripotent transcription factor OCT4 and Nanog, with higher mRNA and protein level. However, Sox2 seems only increased protein level after the expression of PRMT8.
According to previous studies, Sox2 can increase the expression of PRMT8 gene, while pluripotent transcription factors can interact with each other by activating promoter to increase expression [6,14]. Combining with the research results, although PRMT8 genes didn't further promote Sox2 expressing to form a loop, it could increase Sox2 protein content. It meant that PRMT8 may increase Sox2 protein content by methylating on Sox2 to reduce Sox2 degradation, so that Sox2 further raises other pluripotent transcription factors Oct4 and Nanog expression. Therefore, PRMT8 can promote the stemness characteristics of colon cancer cells.
In summary, we showed for the first time that PRMT8 plays an important role in colon cancer cell and it may be a colon cancer treatment target in the future, especially for tumors with chemotherapy resistance. However, PRMT8 may not be the only key point leading to stemness formation in colon cancer cells. In the further mechanism studies, we will further the analysis of interacting protein of PRMT8.
Acknowledgements
This study was supported by grants from the Beijing TCM science and technology development fund project (No. JJ2016-16, to Bangwei Cao; QN2015-08, Jing Wang), Beijing Municipal Natural Science Foundation (No. 7172081, to Bangwei Cao).
Contributions
L.H.S. and C. B. W. designed the study. The data were analyzed by L.H.S., W.B, Y. J., W. J. The figures were prepared by L.H.S. and W.B. Q. L. wrote the paper. All authors reviewed the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. GBD 2015 Risk Factors Collaborators. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1659-724
2. Calley.L.Hirsch Jeffrey L.Wrana, Sharon Y.R. KATapulting toward pluripotency and cancer. Journal of Molecular Biology. 2016;429:1958-77
3. Tan Boon Toh, Jhin Jieh Lim, Edward Kai-Hua Chow. Epigenetics in cancer stem cells. Moleculor Cancer. 2016;16:29-37
4. David L Marks, Rachel LO Olson, Martin E Fernandez-Zapico. Epigenetic control of the tumor microenvironment. Epigenomics. 2016;8:1671-87
5. Oliveira L.R, Jeffrey S.S, Ribeiro-Silva A. Stem cells in human breast cancer. Histol Histopathol. 2010;25:371-85
6. Leis O, Eguiara A, Lopez-Arribillaga E. et al. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012;31:1354-65
7. J. Tang, A. Frankel, R.J. Cook, et al. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J Biol Chem. 2000;275:7723-30
8. Claudia Solari, Camila Vazquez Echegaray, Carlos Luzzani. et al. Protein arginine Methyltransferase 8 gene is expressed in pluripotent stem cells and its expression is modulated by the transcription factor Sox2. Biochemical and Biophysical Research Communications. 2016;473:194-9
9. Jeong H.C, Park S.J, Choi J.J. et al. PRMT8 Controls the Pluripotency and Mesodermal Fate of Human Embryonic Stem Cells by Enhancing the PI3K/AKT/SOX2 Axis. Stem Cells. 2017;35:2037-49
10. Hernandez S, Dominko T. Novel Protein Arginine Methyltransferase 8 Isoform Is Essential for Cell Proliferation. J Cell Biochem. 2016;117:2056-66
11. Alexandra Chittka, Justyna Nitarska, Ursula Grazini. et al. Transcription Factor Positive Regulatory Domain 4 (PRDM4) Recruits Protein Arginine Methyltransferase 5 (PRMT5) to Mediate Histone Arginine Methylation and Control Neural Stem Cell Proliferation and Differentiation. J Biol Chem. 2012;287:42995-3006
12. Nicole Stopa, Jocelyn E. Krebs, David Shechter. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci. 2015;72:2041-59
13. Hunain Alam, Bingnan Gu, Min Gyu Lee. Histone methylation modifiers in cellular signaling pathways. Cell Mol Life Sci. 2015;72:4577-92
14. Metivier R, Penot G, Hubner M.R. et al. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751-63
15. Frietze S, Lupien M, Silver P.A. et al. CARM1 regulates estrogen-stimulated breast cancer growth through upregulation of E2F1. Cancer Res. 2008;68:301-6
16. Wei H, Wang B, Miyagi M. et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF-kappaB. Proc Natl Acad Sci. 2013;110:13516-21
17. Hiroki Matsuda, Yun-Bo Shi. An essential and evolutionarily conserved role of protein arginine methyltransferase 1 for adult intestinal stem cells during postembryonic development. Stem Cells. 2010;28:2073-83
18. Wei H, Mundade R, Lange K.C. et al. Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle. 2014;13:32-41
19. Ding S.L, Yang Z.W, Wang J. et al. Integrative analysis of aberrant Wnt signaling in hepatitis B virus-related hepatocellular carcinoma. World J Gastroenterol. 2015;21:6317-28
20. Fei Q, Shang K, Zhang J. et al. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat Commun. 2015;6:8651-74
21. Zhang H, Weng X, Ye J. et al. Promoter hypermethylation of TERT is associated with hepatocellular carcinoma in the Han Chinese population. Clin Res Hepatol Gastroenterol. 2015;39:600-9
22. Wang W, Ma X.P, Shi Z. et al. Epidermal growth factor receptor pathway polymorphisms and the prognosis of hepatocellular carcinoma. Am J Cancer Res. 2015;5:396-410
23. Gao J, Aksoy B.A, Dogrusoz U. et al. Integrative analysis of complex cancer genomics and clinical profles using the cBioPortal. Sci Signal. 2013;6:l1-9
Author contact
![]() Corresponding author: : Dr. Bangwei Cao, Department of Cancer Center, Beijing Friendship Hospital, Capital Medical University, 95 Yong An Road, Xicheng District, Beijing 100050, China, TEL: +86-010-63139321, Fax: +86-010-63139321, E-mail address: oncologyedu.cn.
Corresponding author: : Dr. Bangwei Cao, Department of Cancer Center, Beijing Friendship Hospital, Capital Medical University, 95 Yong An Road, Xicheng District, Beijing 100050, China, TEL: +86-010-63139321, Fax: +86-010-63139321, E-mail address: oncologyedu.cn.
Received 2017-11-13
Accepted 2018-2-25
Published 2018-4-6
Citation styles
APA
Lin, H., Wang, B., Yu, J., Wang, J., Li, Q., Cao, B. (2018). Protein arginine methyltransferase 8 gene enhances the colon cancer stem cell (CSC) function by upregulating the pluripotency transcription factor. Journal of Cancer, 9(8), 1394-1402. https://doi.org/10.7150/jca.23835.
ACS
Lin, H.; Wang, B.; Yu, J.; Wang, J.; Li, Q.; Cao, B. Protein arginine methyltransferase 8 gene enhances the colon cancer stem cell (CSC) function by upregulating the pluripotency transcription factor. J. Cancer 2018, 9 (8), 1394-1402. DOI: 10.7150/jca.23835.
NLM
Lin H, Wang B, Yu J, Wang J, Li Q, Cao B. Protein arginine methyltransferase 8 gene enhances the colon cancer stem cell (CSC) function by upregulating the pluripotency transcription factor. J Cancer 2018; 9(8):1394-1402. doi:10.7150/jca.23835. https://www.jcancer.org/v09p1394.htm
CSE
Lin H, Wang B, Yu J, Wang J, Li Q, Cao B. 2018. Protein arginine methyltransferase 8 gene enhances the colon cancer stem cell (CSC) function by upregulating the pluripotency transcription factor. J Cancer. 9(8):1394-1402.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.