

Abstract
Neuropathy is the most common and debilitating complication of diabetes and results in pain, decreased motility, and amputation. Diabetic neuropathy encompasses a variety of forms whose impact ranges from discomfort to death. Hyperglycemia induces oxidative stress in diabetic neurons and results in activation of multiple biochemical pathways. These activated pathways are a major source of damage and are potential therapeutic targets in diabetic neuropathy. Though therapies are available to alleviate the symptoms of diabetic neuropathy, few options are available to eliminate the root causes. The immense physical, psychological, and economic cost of diabetic neuropathy underscores the need for causally targeted therapies. This review covers the pathology, epidemiology, biochemical pathways, and prevention of diabetic neuropathy, as well as discusses current symptomatic and causal therapies and novel approaches to identify therapeutic targets.
1. Introduction
Neuropathy is a common and costly complication of both type 1 (T1DM) and type 2 diabetes (T2DM). The prevalence of neuropathy is estimated to be about 8% in newly diagnosed patients and greater than 50% in patients with longstanding disease (Boulton, 2005). There is increasing evidence that even pre-diabetic conditions are also associated with some forms of neuropathy (Franklin, 1990; Singleton, 2003). An estimated 15% of all patients with diabetes will develop foot ulcers (Gordois, 2003), and diabetic neuropathy is the leading cause of nontraumatic limb amputation (Thomas, 1999). The annual costs of diabetic neuropathy and its associated morbidities in the US have been estimated to exceed $10.9 billion (Gordois, 2003).
In recent years, considerable progress has been made toward understanding the biochemical mechanisms leading to diabetic neuropathy, and as a result, new treatment modalities are being explored. This review will discuss the epidemiology and impact of diabetic neuropathy and the current understanding of its pathogenesis. This will be followed by a discussion of the diagnosis and evaluation of diabetic neuropathy, and conclude with an examination of current treatment options and anticipated new therapeutic approaches. Definition. Diabetic neuropathy is a descriptive term that encompasses a spectrum of clinical and subclinical syndromes with differing anatomical distributions, clinical courses, and possibly differing underlying pathogenetic mechanisms. Each is characterized by diffuse or focal damage to peripheral somatic or autonomic nerve fibers resulting from diabetes mellitus, although indistinguishable syndromes may occur idiopathically or in association with other disorders in nondiabetic individuals. Table 1 lists the most common clinical syndromes comprising diabetic neuropathy.
Table 1.
Classification of Clinical Diabetic Neuropathy
| Diffuse Neuropathy | Focal Neuropathy |
|---|---|
Distal symmetrical sensorimotor polyneuropathy (DPN)
Diabetic autonomic neuropathy (DAN)
|
Mononeuropathy Mononeuropathy multiplex Plexopathy Radiculopathy Cranial neuropathy |
The syndromes may be grouped under two general headings: diffuse and focal neuropathies. The diffuse neuropathies, i.e., distal symmetrical sensorimotor polyneuropathy (DPN) and diabetic autonomic neuropathy (DAN) are common, usually chronic, and often progressive. The focal neuropathies are less common, usually acute in onset, and often self-limited.
In DPN, sensory deficits usually overshadow motor nerve dysfunction and appear first in the distal portions of the extremities and progress proximally in a “stocking-glove” distribution with increasing duration or severity of diabetes [Figure 1]. The signs and symptoms of DPN vary depending on fiber type involved, with large fiber disease impairing proprioception and light touch. Small fiber disease impairs pain and temperature perception, leading to paresthesias, dysesthesias, and/or neuropathic pain. Distal weakness occurs only in the most severe cases. Diminished or absent deep-tendon reflexes, particularly the Achilles tendon reflex, often indicates mild and otherwise asymptomatic DPN. More advanced asymptomatic neuropathy may first present with late complications such as ulceration or neuroarthropathy (Charcot’s joints) of the foot.
Figure 1. Stocking Glove Configuration of DPN.

Diabetic neuropathy is dependent on axon length, initiating in the toes and progressing upward until reaching the calf. Neuropathy presents at the fingertips at this point.
Diabetic autonomic neuropathy (DAN) is the other form of diffuse diabetic neuropathy. DAN often accompanies DPN and, as detailed in Table 2, can impair any sympathetic or parasympathetic autonomic function. Although DAN is highly prevalent and associated with a markedly reduced quality of life and increased mortality, it is among the least recognized and most poorly understood complications of diabetes (Freeman, 2005). Further, many of the clinical symptoms of DAN are common and may be due to causes other than diabetic neuropathy. Therefore, as will be discussed in a subsequent section on diagnosis and treatment, it is important to rule out non-diabetes related etiologies for the specific symptom(s) a diabetic patient is experiencing before making a final diagnosis.
Table 2.
Clinical Manifestations of Diabetic Autonomic Neuropathy
|
|
| - |
|
|
|
The focal forms of diabetic neuropathy reflect damage to single (mononeuropathy) or multiple peripheral nerves (mononeuropathy multiplex), cranial nerves, regions of the brachial or lumbosacral plexuses (plexopathy), or the nerve roots (radiculopathy). The most common peripheral nerve mononeuropathies, medial and ulnar neuropathy, are essentially indistinguishable from entrapment neuropathies in nondiabetic subjects, suggesting that the diabetic nerve has increased susceptibility to compression. Indeed, carpal tunnel syndrome can be demonstrated electrophysiologically in 20 to 30% of diabetic patients, and presents as a clinically relevant problem in 5 to 10% of patients with diabetes (Dyck, 1993). Except for these mononeuropathies, the focal diabetic neuropathies are relatively uncommon, acute in onset, self-limiting, and tend to occur in older patients. The most common cranial neuropathy affects the third nerve, producing unilateral headache, diplopia, and ptosis with pupillary sparing (diabetic ophthalmoplegia) (Vinik, 2004b). Both lumbosacral plexopathy and polyradiculopathy, sometimes referred to as diabetic amyotrophy, occur in diabetic patients, particularly elderly males with T2DM. These conditions usually present with pain, particulary of one thigh, involve L2 thru L4 innervated muscles and self resolve over time. Thoracic radiculopathy produces unilateral band like or abdominal pain that may be misdiagnosed as an acute intrathoracic or intra-abdominal emergency.
In summary, DPN and DAN are very common, generally diffuse, and progressive. The focal neuropathies, with the exception of median and ulnar neuropathies, are generally rare, sudden in onset, often self-limited, and tend to occur in older patients. The remainder of this module will focus on DPN and DAN. The reader is referred to a review by Vinik and colleagues for an extensive discussion of the focal neuropathies(Vinik, 2004a).
2. Epidemiology and Impact of Diabetic Neuropathy
Diabetic Polyneuropathy (DPN)
Estimating the prevalence, incidence, and risk of DPN depends on the criteria employed to identify the syndrome. The American Academy of Neurology, the American Association of Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation published a consensus definition in 2005. Important points in the case definition include: 1) the combination of neuropathic symptoms, signs and abnormal electrodiagnostic studies is the strongest predictor of DPN, 2) symptoms alone are a poor predictor of disease, 3) electrodiagnostic studies are not required for the clinical definition of DPN, but are recommended to monitor disease in clinical research protocols(England, 2005). Thus, the definition of DPN may include a symptom score, a focused neurological examination and nerve conduction studies, or, optimally, all of these. Yet, regardless of the diagnostic criteria, it is clear that 1) DPN is highly prevalent in patients with diabetes, 2) its prevalence increases with the duration of diabetes and 3) strict glycemic control reduces the incidence and progression of diabetic neuropathy. Table 3 summarizes several reports on the prevalence of DPN in diabetic patients.
Table 3.
Reported Prevalence of Diabetic Peripheral Neuropathy
| Reference | Diagnostic Criteria | Initial Prevalence (%) | Time of Initial Assessment | Later Prevalence (%) | Time of Later Assessment |
|---|---|---|---|---|---|
|
| |||||
| (Pirart, 1978) | Neurological examination | 7.5 | At diagnosis | 50 | 25 yr post-diagnosis |
|
| |||||
| (Palumbo, 1978) T2DM | Symptoms and/or decreased vibratory sense | 4 | Within 5 yrs of diagnosis | 15 | 20 yr post-diagnosis |
|
| |||||
| (Young, 1993) T1DM. T2DM | Neurological examination and symptoms score | 20.8 | 36.8 | > 10 yr | |
|
| |||||
| (Dyck, 1993) T1DM, | Two or more abnormalities (symptoms, nerve conduction, QST*, AFT**) | T1DM: 54 | Mean 14.5 yr | ||
| T2DM | T2DM: 45 | Mean 8.1 yr | |||
|
| |||||
| (Partanen, 1995)T2DM | Probable polyneuropathy | 8.3 | At Diagnosis | 41.9 | 10 yr post-diagnosis |
| Absent reflex | 28 | 46 | |||
|
| |||||
| (The effect of intensive diabetes therapy on the development and progression of neuropathy. The Diabetes Control and Complications Trial Research Group, 1995)DCCT-T1DM | Confirmed clinical neuropathy | 2.1 | Baseline (diabetes duration 1–5 yr) | 9.6 | 5 yr post-baseline |
| Abnormal nerve conduction | 21.8 | 40.2 | |||
|
| |||||
| (Tesfaye, 1996)T1DM | ≥ 2 criteria | 12 | <7 yr duration | 42 | ≥ 15 yr duration |
QST: quantitative sensory tests;
AFT: autonomic function tests
The prevalence of DPN increases with age, and tends to be more common in patients with T2DM than in those with T1DM. Figure 2 illustrates the prevalence of DPN as a function of disease duration and age interval observed in a cross-sectional study of 6,487 patients (~ 37% with T1DM). In the overall population the prevalence of DPN was significantly higher in patients with T2DM (32.1%) than in T1DM patients (22.7%, P < 0.001) and there was a highly-significant correlation between age and prevalence of neuropathy in both T1DM and T2DM (Young, 1993).
Figure 2. Effect of Disease Duration and Age on Prevalence of Diabetic Neuropathy.

For both T1DM and T2DM, duration of diabetes (left) as well as age of patient (right) is correlated to the incidence of diabetic neuropathy. Adapted from (Young, 1993)
Similar rates of DPN were reported in the Rochester Diabetic Neuropathy Study (Dyck, 1993). In 1986, 380 patients were enrolled in this study; 102 (26.8%) had T1DM and 278 (73.2%) had T2DM. Patients were assessed for DPN by sign and symptom scores coupled to physiological assessments of nerve function, including nerve conduction studies and quantitative sensory testing. 54% of patients with TIDM with average disease duration of 14.5 years had DPN, while 45% of T2DM patients with average disease duration of 8.1 years had DPN.
The EURODIAB study examined 3,250 T1DM patients from 16 European countries. DPN was defined as the presence of 2 or more abnormalities in either symptoms, signs, quantitative sensory or autonomic function testing. The prevalence of DPN across Europe was 28%, with a strong correlation between duration of diabetes and level of glycemic control (Tesfaye, 1996).
The Diabetes Control and Complications Trial (DCCT) demonstrated that intensive treatment in patients with T1DM significantly reduced both the incidence (The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group, 1993) and progression (The effect of intensive diabetes therapy on the development and progression of neuropathy. The Diabetes Control and Complications Trial Research Group, 1995) of neuropathy. Figure 3 depicts the prevalence of neuropathy in the primary prevention cohort of the DCCT after 5 years treatment with either a conventional or intensive insulin regimen in the DCCT.
Figure 3. Effect of Glycemic Control on Diabetic Neuropathy in DCCT.

Intensive therapy cohort which showed better glycemic control, results in lower incidences of all forms of diabetic neuropathy compared to conventional therapy. Adapted from (The effect of intensive diabetes therapy on the development and progression of neuropathy. The Diabetes Control and Complications Trial Research Group, 1995)
HbA1c levels in the intensive and conventional treatment groups were separated by about 2 percentage points throughout the follow-up (7.2% vs. 9.1%, respectively). Intensive treatment was associated with a 71% risk reduction for confirmed clinical neuropathy (abnormal history, physical examination, or both, confirmed by abnormal nerve conduction or abnormal autonomic function tests), a 54% risk reduction for clinical neuropathy, a 59% risk reduction for abnormal nerve conductions, and a 56% risk reduction for autonomic nervous system dysfunction. This figure also highlights the difference in prevalence estimates related to the stringency of criteria employed, as well as the lower prevalence of autonomic vs. peripheral neuropathy early in the disease.
In the United Kingdom Prospective Diabetes Study, 3,867 newly diagnosed T2DM patients were randomized into either intensive treatment with an oral hypoglycemic agent or insulin or conventional treatment with diet. After 10 years, intensive treatment resulted in approximately 1% lower HbA1c vs. conventional treatment and was associated with a 25% risk reduction in microvascular endpoints (retinopathy, nephropathy and neuropathy)(Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group, 1998). However, most of this reduction was due to fewer patients in the intensive treatment arm requiring retinal photocoagulation compared to patients in the conventional arm. While there was a tendency toward a reduction in death from peripheral vascular disease and the prevalence of amputation, effects on these single endpoints failed to achieve statistical significance. Similarly, surrogate endpoints for neuropathy showed trends toward improvement in the intensive treatment group at year 10 and in a smaller group of patients followed for 15 years, the prevalence of impaired sensory perception in the lower extremities was significantly preserved by intensive versus conventional treatment (31.2% vs. 51.7%, P = 0.0052).
Height is another risk factor for DPN, suggesting that longer fibers are more vulnerable to injury. Other suggested risk factors for DPN include smoking (Tesfaye, 1996), excessive alcohol use (Adler, 1997), hypertension (Forrest, 1997), low plasma insulin levels (Partanen, 1995), and co-morbid diabetic complications (Cohen, 1998).
The most common morbidities related to DPN are recurrent foot infections, ulcers and amputations, and Charcot’s joints. It was estimated that upwards of 15% of patients with diabetes will develop at least one foot ulcer (Boulton, 2004b), and one recent study observed an annual incidence of nearly 2% (Ramsey, 1999). While vascular disease and ischemia contribute, it has been reported that 60 to 70% of diabetic foot ulcers are neuropathic in origin (Gonzalez, 2000). A significant proportion of neuropathic diabetic foot ulcers is accompanied by cellulitis or osteomyelitis (about 15%) and these conditions contribute to the annual incidence of lower extremity amputation in patients with diabetes, which has been estimated to be around 0.6% (van Houtum, 2004). In 1999, the attributable cost for a 40- to 65-year old man with a new foot ulcer was estimated to be $28,000 for the 2 years after diagnosis (Ramsey, 1999) and in 2003, the total annual cost of DPN and its complications in the US was estimated to be between $4.6 and $13.7 billion – representing up to 27% of the direct medical cost of diabetes (Gordois, 2003). Furthermore, there are important quality of life issues for patients with DPN including pain and other forms of discomfort, decreased mobility, and a variety of psychosocial impairments (Vileikyte, 2003).
There are relatively few data available regarding the influence of DPN on mortality. However, in their study of diabetic foot ulcers, Ramsey and colleagues found that 3-year survival in patients with foot ulcers was ~ 17% less than in age- and sex-matched diabetic patients (Ramsey, 1999). Further, using a newly-developed accelerated failure time model which included alcohol consumption, proteinuria, race, retinopathy, sex, smoking, type of diabetes, BMI, duration of diabetes, HbA1c, and “toescore” (an age-adjusted transformation of vibration perception threshold), it was found that toescore was the most significant contributor to mortality (Coppini, 2000). Thus, it is clear that DPN exacts an enormous toll.
Diabetic Autonomic Neuropathy (DAN)
Despite its negative impact on survival and quality of life in persons with diabetes, DAN remains poorly understood. Since all organs receive input from the autonomic nervous system (ANS), DAN can affect every body system. Most organs are dually innervated, receiving fibers from the parasympathetic and the sympathetic branches of the ANS. Because the vagus nerve (parasympathetic), the longest autonomic nerve, mediates approximately 75% of all parasympathetic activity (Low, 2004) and neuropathy is seen first in the longest fibers, the earliest manifestations of DAN tend to be parasympathetic and are usually widespread.
Although clinical symptoms of DAN may not appear until long after diabetes onset, subclinical neuropathy may be detected within a year of diagnosis in T2DM and two years of diagnosis in T1DM (Pfeifer, 1984). Such findings highlight the importance of screening for DAN. As for DPN, the reported prevalence varies greatly depending on the criteria used to identify DAN as well as the population studied. These range from as low as 2.5% of the primary prevention cohort in the DCCT (retinopathy and microalbuminuria-free patients with T1DM of 1 to 5 years duration) (The effect of intensive diabetes therapy on the development and progression of neuropathy. The Diabetes Control and Complications Trial Research Group, 1995) to as high as 90% of patients with long-standing T1DM who were potential candidates for a pancreas transplant (Kennedy, 1995).
In 1988 the San Antonio Conference on Diabetic Neuropathy provided guidelines for standardized objective measurements of neuropathy based on clinical symptoms, clinical examination, electrodiagnostic studies (EDX), quantitative sensory testing (QST), and autonomic function testing for use in clinical research studies. It was further recommended that these measures be standardized to a normative control population (Asbury, 1988), and since that time, several studies have assessed the prevalence of DAN in defined populations. Like DPN, the diagnosis of DAN depends on specific clinical and physiological assessments. Most commonly, changes in heart rate with deep breathing, position change (lying to standing), and breathing out against pressure (Valsalva ratio) are measured in each patient. EKG leads are applied to the chest and the EKG recorded briefly while the patient inhales and exhales slowly and regularly 6 times per minute (E/I ratio, normal value ≥ 1.04), stands (30:15 ratio, normal value ≥ 1.08), or blows out against a closed tube (Valsalva ratio, normal value ≥ 1.26). Thus while DAN can involve many body systems, the cardiovascular system is most commonly tested, and frequently measures of DAN are, in reality, measures of cardiac autonomic neuropathy. In a community-based study, the overall prevalence of DAN assessed by heart rate variability was 16.7%, being somewhat more common in patients with T1DM (20.9%) than in those with T2DM (15.8%). Symptomatic DAN was much less common (2.5%) than that detected by autonomic function tests, but was significantly more frequent in patients with T1DM (5/43, 11.6%) than in patients with T2DM (1/202, 0.5%).
In a larger study (647 patients with T1DM, 524 patients with T2DM), Ziegler and colleagues defined DAN as an abnormality of ≥ 2 of 6 autonomic function tests. In addition to the 3 more standard tests discussed above, a spectral analysis of the EKG in the low- and mid-frequency bands and a vector analysis of deep breathing were performed to complete the battery of autonomic function tests. The investigators found that 25.3% of patients with T1DM and 34.3% of those with T2DM had abnormal findings in ≥ 2 of 6 autonomic function tests. If more restrictive criteria were used for diagnosis (≥ 3 of 6 abnormal autonomic function tests), the prevalence of autonomic neuropathy was 16.8% and 22.1% for patients with T1DM and T2DM (Ziegler, 1993). In another study of 110 children and adolescents with T1DM, abnormality of one or more of the 3 standard autonomic function tests was found in 42.7% of patients (Verrotti, 1995). In summary, DAN is a common form of neuropathy in diabetic patients. There is no convincing evidence for a difference in prevalence between T1DM and T2DM, but similar to DPN, the prevalence is highly dependent on the criteria used to define DAN.
Similar to DPN, the risk of DAN increases with inadequate glycemic control and with disease duration. For example, in the Appropriate Blood pressure Control in Diabetes trial, assessment of DAN was made in 869 patients with T2DM after washout of previous antihypertensive treatment. Stepwise logistic regression analysis of potential risk factors revealed that HbA1c and duration of diabetes were independently associated with DAN (Cohen, 1998). The Diabetic Cardiovascular Autonomic Neuropathy study examined the potential clinical correlates of cardiac autonomic neuropathy in 647 patients with T1DM and in 524 patients with T2DM. Stepwise regression analysis showed a significant association of cardiac autonomic neuropathy and HbA1c in patients with T1DM, but not in those with T2DM (Ziegler, 1993). Other risk factors or significant correlates of DAN that have been reported include hypertension, female gender, LDL-cholesterol, HDL-cholesterol in patients with T1DM (Maser, 1990), retinopathy, DPN in patients with T1DM or T2DM (Ziegler, 1993), and albuminuria in patients with T2DM (Cohen, 1998).
Due to the importance of the autonomic nervous system in regulating virtually every body function, the consequences of DAN are many and varied, ranging from bothersome, to debilitating, to deadly. Cardiac autonomic neuropathy (CAN) is the most clinically important manifestation of DAN due to its association with several negative outcomes, including increased mortality. Early markers of CAN include resting tachycardia and loss of heart rate variation during deep breathing, whereas loss of heart rate response to mild exercise is indicative of nearly complete cardiac denervation. Impaired parasympathetic function causes loss of bradycardic responses to sleep and to deep inspiration. Impaired sympathetic function, which generally occurs as the syndrome progresses, can increase cardiac adrenergic sensitivity, which may predispose a patient to tachycardia and sudden death. A prolonged corrected QT interval (QTc) indicates an imbalance between right and left sympathetic innervation may increase risk for arrhythmias. Other common and potentially dangerous manifestations of cardiac autonomic neuropathy include exercise intolerance, orthostatic hypotension, and intraoperative cardiovascular lability (Vinik, 2003).
Another important syndrome associated with CAN is silent myocardial ischemia or “cardiac denervation syndrome.” Reduced appreciation of ischemic pain can impair timely recognition of myocardial ischemia or infarction and delay appropriate treatment. Many studies have compared the prevalence of silent myocardial ischemia, usually measured by exercise stress testing, in diabetic patients with and without CAN. Collectively these studies show cardiac autonomic neuropathy greatly increases the risk of silent myocardial ischemia. Conclusions from a large body of literature on cardiac autonomic neuropathy and various other diabetic cardiovascular morbidities make it abundantly clear that the presence of CAN greatly increases the risk of all-cause mortality, cardiovascular mortality, and major cardiovascular events, with relative risks ranging from 2 to > 4 (Freeman, 2005). There is also often an association between CAN and diabetic nephropathy, suggesting other co-morbidities likely contribute to the increased cardiovascular risk (Maser, 2003).
The most devastating outcome of DAN is the excess mortality associated with CAN. Many studies have explored the potential contribution of CAN to total mortality in patients with diabetes – both T1DM and T2DM. Of 15 studies included in a meta-analysis by Maser and colleagues (Maser, 2003), 14 reported increased mortality rates in patients with CAN; in those studies the risk ratio for all cause mortality in patients with CAN vs. those without CAN at baseline (follow-up = 0.5 to 16 years) ranged from 2.1 (Sawicki, 1996) to 9.2 (Jermendy, 1991). A statistically significant increase in mortality was reported in 12 of the studies.
As a whole there was a strong and consistent association between CAN and increased risk of mortality, however the association was much stronger (RR = 3.45, P < 0.001) in 10 studies where CAN was identified using 2 or more autonomic function tests than in studies that used a single test (RR = 1.20, P = 0.03). This difference could reflect the presence of more severe autonomic dysfunction in patients with CAN identified with multiple tests or a higher prevalence of co-morbid conditions. Alternatively, use of a single abnormal autonomic function test to identify CAN could allow a higher frequency of misclassification. Nonetheless, it may be concluded that CAN greatly increases the risk of mortality. Although symptoms of CAN may not appear until relatively late in disease progression, autonomic neuropathy has a profound impact, and regular screening for CAN in all patients with diabetes is advisable.
Gastrointestinal manifestations of DAN are diverse and can affect any portion of the gastrointestinal tract. Esophageal dysfunction resulting from vagal neuropathy may cause heartburn and dysphagia for solids (Freeman, 2005). Delayed gastric emptying (gastroparesis) was reported to occur in approximately 50% of patients with longstanding diabetes (Kong, 1999). Although diabetic gastroparesis is usually relatively benign, severe cases may cause nausea, vomiting, epigastric discomfort, and bloating which can last for extended periods or occur in cycles. Diarrhea, with or without intermittent constipation, is another common and often debilitating symptom of DAN (Low, 2004).
Genitourinary manifestations of DAN can include increased or decreased urinary frequency, bladder over distention, and urine retention or overflow incontinence. Patients with bladder dysfunction are predisposed to developing urinary tract infections, which may accelerate or exacerbate renal failure. Erectile dysfunction and retrograde ejaculation in men and decreased libido and decreased vaginal lubrication in women are also common and early findings in patients with DAN (Freeman, 2005; Low, 2004).
Autonomic sudomotor dysfunction can produce distal anhydrosis with a stocking-glove distribution similar to that of DPN. Although this manifestation of DAN is essentially asymptomatic, it can predispose a patient to heatstroke and hyperthermia and may produce a compensatory central hyperhydrosis (Freeman, 2005).
3. Clinical Evaluation of Diabetic Neuropathy
A consensus statement from the San Antonio Conference on Diabetic Neuropathy recommended that the diagnosis and classification of DPN for research and clinical trials be based on at least one standardized measure from each of the following categories: clinical symptoms, clinical examination, EDX, QST, and AFT (Ziegler, 1993). Many currently-used techniques are based on methods developed by Dyck and colleagues at the Mayo Clinic (Dyck, 1992). In the absence of neurological symptoms or clinically detectable neurological deficits indicative of a diffuse or focal neuropathy, subclinical neuropathy can be diagnosed and staged as outlined in Table 4. It is important to note that the diagnosis of subclinical or clinical DPN requires that signs (e.g., abnormal quantitative tests for subclinical neuropathy) and symptoms (for clinical neuropathy) are not attributable to a nondiabetic etiology. Because there are no distinguishing features unique to diabetic neuropathy, all other possible causes of the observed neuropathic disorders must be ruled out by careful history and physical examination.
Table 4.
Classification and Staging of Diabetic Neuropathy
| Class I: Subclinical Neuropathy | |
|
| |
Abnormal Electrodiagnostic Tests
|
Class Ia: normal EDX, abnormal AFT or QST |
|
| |
Abnormal Quantitative Sensory Testing
|
Class Ib: abnormal EDX or abnormal AFT, and QST |
|
| |
Abnormal Autonomic Function Tests
|
Class Ic: abnormal EDX and either abnormal AFT or QST, or both |
|
| |
| Class II: Clinical Neuropathy – as detailed in Table 1 | |
|
| |
| AFT: autonomic function test | |
| EDX: electrodiagnostic tests, eg, nerve conduction and evoked potential | |
| QST: quantitative sensory testing | |
A simpler approach may be taken in an outpatient setting. In a recent statement by the American Diabetes Association, DPN is defined as “the presence of symptoms and/or signs of peripheral nerve dysfunction in people with diabetes after the exclusion of other causes” (Boulton, 2005). The diagnosis requires a careful history and clinical examination of the feet. Several instruments for this purpose have been developed and validated. One example is the Michigan Neuropathy Screening Instrument (MNSI) (Feldman, 1994), shown in Figure 4. This patient questionnaire consists of 15 questions about sensation, general asthenia, and peripheral vascular disease. A positive response on ≥ 7 of the questions is diagnostic of diabetic peripheral neuropathy and correlates well with neuropathy diagnosed by the Mayo Clinic criteria.
Figure 4. MNSI Patient Questionnaire.

Adapted with permission
The MNSI questionnaire is followed by a simple 8-point clinical examination involving inspection of the foot, assessment of ankle reflexes, and semi quantitative determination of vibration perception (Figure 5). An MNSI score > 2 indicates the presence of neuropathy with a high specificity (~ 95%) and sensitivity (~ 80%) (Bax, 1996).
Figure 5. 8 Point Clinical Examination for Diabetic Neuropathy.

Adapted with permission
Patients with an abnormal MNSI score may undergo a more detailed examination such as that summarized by the Michigan Diabetic Neuropathy Score (MDNS), in which the severity of neuropathy is determined through a focused, 46-point neurological examination that includes measures of sensory impairment, muscle strength, and reflexes (Feldman, 1994). Other more complicated techniques to assess warm and cold perception thresholds, current perception thresholds, etc, have also been developed for research purposes. These are generally time-consuming and require specialized equipment, and thus are not routinely employed in a clinic setting.
Nerve conduction studies can be used to quantify the degree of nerve injury in DPN (Boulton, 2004c). While not usually required for the diagnosis, nerve conduction studies can help the patient and physician monitor DPN progression over a long period of time, particularly if the patient is asymptomatic. Nerve conduction studies are also useful to identify superimposed mononeuropathies, e.g., carpal tunnel syndrome. These superimposed mononeuropathies are a common problem in patients with DPN.
As for DPN, DAN is usually classified as either subclinical or clinical depending upon the presence of clinical manifestations of autonomic dysfunction. As mentioned previously, the clinical manifestations of DAN can be many and varied, but may not occur until long after DAN can be detected through autonomic function tests. Routinely, five simple, noninvasive cardiovascular reflex tests are used for diagnosis (partly described earlier): Valsalva maneuver, heart rate response to deep breathing, heart rate response to standing up, blood pressure response to standing up, and blood pressure response to sustained handgrip (Freeman, 2005). Abnormalities in these noninvasive tests of cardiovascular function show a strong correlation with symptoms (Low, 2004), and with autonomic dysfunction in other organs including pupillomotor function, gastrointestinal function, and norepinephrine production (Freeman, 2005; Vinik & Mehrabyan, 2004b). However, abnormalities in lower-extremity sudomotor function and impotence may precede detectable impairment in the simple cardiovascular autonomic function tests (Freeman, 2005; Low, 1986; Vinik & Mehrabyan, 2004b). Table 5 lists several noninvasive tests of cardiovascular autonomic function and diagnostic values for CAN (Freeman, 2005).
Table 5.
Diagnostic Tests for Cardiovascular Autonomic Neuropathy
| Diagnostic Tests of CV Autonomic Neuropathy | |
|---|---|
|
| |
| Test | Diagnostic Value |
| Resting heart rate | >100 bpm |
|
| |
| Beat-to-beat HR variation (HRV) | |
| Abstain from coffee overnight | |
| Do not test after hypoglycemic episode | |
| Supine position, 6 breaths per minute | Difference < 10 bpm or expiration:inspiration R-R ratio > 1.17 |
|
| |
| Heart rate response to standing | |
| R-R interval measured at beats 15 and 30 after standing (normally tachycardia is followed by reflex bradycardia) | 30:15 ratio > 1.03 |
|
| |
| Heart rate response to Valsalva maneuver | |
| Patient forcibly exhales into manometer mouthpiece, exerting 40 mmHg pressure for 15 seconds | Ratio of longest to shortest R-R interval < 1.2 |
|
| |
| Systolic BP response to standing | |
| Measure in supine position and 2 minutes after standing | Decrease > 30 mmHg (10 to 29 is borderline) |
|
| |
| Diastolic BP response to isometric exercise | |
| Establish patient’s maximum handgrip pressure | |
| Exert 30% maximum for 5 minutes | Increase < 16 mmHg in contralateral arm |
|
| |
| Electrocardiography | QTc > 440 ms |
Resting tachycardia and loss of heart rate variation in response to breathing or the Valsalva maneuver are primary indicators of parasympathetic dysfunction and are among the earliest signs of CAN. Orthostatic hypotension and loss of blood pressure responses to exercise or handgrip are signs of sympathetic dysfunction and tend to occur later in disease progression. Other more complex tests of cardiovascular autonomic function may be used for research purposes, including spectral analysis of 24-hour heart rate variability, measures of neurovascular flow with Doppler technology, and scintigraphic assessment of cardiovascular sympathetic innervation.
Because many prescription and non-prescription medicines, alcohol, and tobacco can influence autonomic nervous system activity, patients should be tested in an overnight-fasted state, having refrained from alcohol and tobacco for 24 hours, and they should omit even prescription drugs on the day of the test. Further, due to the influence of antecedent hypoglycemia on autonomic function (Dagogo-Jack, 1993), patients should not be tested within 24 hours of a hypoglycemic episode. As with DPN, it is important to rule out possible causes of abnormal AFTs other than DAN through patient history and physical examination. Abnormalities in two or more of the simple AFTs suggest a diagnosis of DAN. In a recent statement by the American Diabetes Association, it is recommended that patients with T1DM be tested five years after diagnosis of diabetes and be tested yearly thereafter. Patients with T2DM should be tested at diagnosis and yearly thereafter (Boulton, 2005).
4. Pathogenesis of Diabetic Neuropathy
There may be multiple etiologies which account for the various neuropathic syndromes seen in patients with diabetes. Hyperglycemia clearly plays a key role in the development and progression of diabetic neuropathy as well as the other microvascular complications of diabetes. Understandably, then, investigations into the molecular and biochemical pathophysiology of diabetic neuropathy have focused on glucose metabolic pathways. Over the past 25 years animal experiments and in vitro studies have identified biochemical pathways likely to be important in the development of diabetic complications and have led to possible approaches to treatment. All of these pathways are related to the metabolic and/or redox state of the cell. Pathways which are mainly driven by metabolism are: glucose flux through the polyol pathway; the hexosamine pathway; excess/inappropriate activation of protein kinase C (PKC) isoforms; accumulation of advanced glycation end-products. While each pathway may be injurious alone, collectively they cause an imbalance in the mitochondrial redox state of the cell and lead to excess formation of reactive oxygen species (ROS) (Kong, 1999; Vinik, 2003) (Figure 4). Increased oxidative stress within the cell leads to activation of the Poly(ADP-ribose) polymerase (PARP) pathway, which regulates the expression of genes involved in promoting inflammatory reactions and neuronal dysfunction. This section will discuss each of these mechanisms and the central role of ROS. This review will not discuss the proposed mechanisms underlying the focal neuropathies. The reader is referred to the review by Vinik and colleagues for this discussion (Vinik, 2004a). Proposed mechanisms underlying DPN and DAN which will not be covered in detail include autoimmune mediation. For a thorough review of this topic, please also see Vinik and colleagues (Calcutt, 2008; Vinik & Mehrabyan, 2004b).
Diabetic neuropathy is thought to occur from both hyperglycemia-induced damage to nerve cells per se and from neuronal ischemia caused by hyperglycemia-induced decreases in neurovascular flow. Much of the basic science addressing the etiology/mechanisms of microvascular complications has used non-neuronal derived cells or cell lines, but studies in animal models of neuropathy, and/or human clinical studies with specific inhibitors of each pathway suggest that each mechanism can contribute to diabetic neuropathy.
4a. Polyol Pathway
The enzyme aldose reductase (AR) reduces glucose to sorbitol and sorbitol dehydrogenase (SDH) oxidizes sorbitol to fructose (Figure 6). Both of these enzymes are abundantly expressed in tissues prone to diabetic complications. Hyperglycemia activates the aldose reductase pathway primarily by mass action: Increased flux through the AR pathway causes increased intracellular sorbitol, a relative intracellular hypertonic state, and compensatory efflux of other osmolytes such as myo-inositol (MI, important in signal transduction) and taurine (an antioxidant) (Nakamura, 1999; Vincent, 2004b). Since NADPH is consumed by aldose reductase-mediated reduction of glucose to sorbitol (Brownlee, 2005; Jermendy, 1991) and NADPH is required for regeneration of reduced glutathione (GSH), this too contributes to oxidative stress. The second step in the polyol pathway oxidizes sorbitol to fructose via sorbitol dehydrogenase (Feldman, 1997). Formation of fructose promotes glycation as well as depletes NADPH, further augmenting redox imbalance. Activation of aldose reductase may also increase formation of diacylglycerol, which activates the deleterious PKC pathway (discussed below) (Uehara, 2004; Yamagishi, 2003).
Figure 6. Schematic of Hyperglycemic Effects on Biochemical Pathways in Diabetic Neuropathy.

Excessive glucose metabolism generates excess NADH and leads to overload of the electron transport chain causing oxidative stress, damage to Mt, activation of PARP. PARP activation by ROS acts in conjunction with the hexosamine and PKC pathway to induce inflammation and neuronal dysfunction. A combination of oxidative stress and hyperglycemia activate the detrimental pathways of AGE, polyol, hexosamine and PKC pathways which lead to redox imbalance, gene expression disturbances, and further oxidative stress. These pathways also induce inflammation and neuronal dysfunction. NF-κB :Nuclear factor kappa B; PARP: Poly(ADP-ribose) polymerase; PKC: Protein kinase C; AGE: Advanced glycation endproducts; RNS: Reactive nitrogen species; ROS: Reactive oxygen species, GSH: glutathione; GSSG: oxidized glutathione; UDPGlcNAc: UDP-N-Acetylglucosamine; VEGF: Vascular endothelial growth factor.
4b. Hexosamine Pathway
In the late 1990’s, the hexosamine pathway was implicated as an additional factor in the pathology of diabetes-induced oxidative stress and complications. Fructose-6 phosphate is a metabolic intermediate of glycolysis. However, during glucose metabolism some fructose-6-phosphate is shunted from the glycolytic pathway to the hexosamine pathway. Here fructose-6 phosphate is converted to glucosamine-6 phosphate by glutamine fructose-6 phosphate amidotransferase (Thornalley, 2005). Glucosamine-6 phosphate is then converted to uridine diphosphate-N-acetyl glucosamine (UDP-GlcNAc), a molecule that attaches to the serine and threonine residues of transcription factors (Brownlee, 2001). Hyperglycemic conditions create additional flux through the hexosamine pathway, ultimately resulting in excess GlcNAc and abnormal modification of gene expression (Brownlee, 2001; Kolm-Litty, 1998; Sayeski, 1996).
Specifically, hyperglycemic conditions and excess GlcNAc cause increased activation of Sp1, a transcription factor implicated in diabetic complications. Sp1 is responsible for the expression of many glucose-induced “housekeeping” genes including transforming growth factor-β1 (TGF-β1) and plasminogen activator inhibitor-1 (PAI-1) (Brownlee, 2001; Du, 2000). Overexpression of TGF-β1 leads to increased collagen matrix production which promotes endothelial fibrosis and decreases proliferation in mesangial cells (Hirakata, 1996; Kolm-Litty, 1998). Overexpression of PAI-1 promotes vascular smooth muscle cell mitosis which plays a role in atherosclerosis (Sayeski & Kudlow, 1996). PAI-1 is not only upregulated via the hexosamine pathway but also the PKC pathway (Figure 6). Thus, two discrete pathways leading to diabetic complications converge through the same injurious mechanism. It has additionally been shown that GlcNAc impairs β-cell function by inducing oxidative stress; increased glutamine fructose-6 phosphate amidotransferase or glucosamine leads to increased hydrogen peroxide levels and reduced expression of insulin, glucose transporter 2, and glucokinase genes (Kaneto, 2001). Thus, increased flux through the hexosamine pathway has been causally implicated in multiple metabolic derangements in diabetes.
4c. PKC Pathway
The protein kinase C (PKC) pathway is an additional mechanism by which hyperglycemia causes injury in complications-prone tissues. Elevated glucose levels stimulate diacylglycerol (DAG), which in turn activates PKC. Increased production of the PKC β-isoform in particular has been implicated in overexpression of the angiogenic protein vascular endothelial growth factor (VEGF), PAI-1, NFκB, TGF-β and the development of diabetic complications such as retinopathy, nephropathy, and cardiovascular disease (Figure 6) (Arikawa, 2007; Das Evcimen, 2007; Veves, 2001). Data on the effects of PKC-β and VEGF on diabetic neuropathy are less clear, but generally most support the concept that increased PKC pathway flux plays a roll in neuropathy as well (Arikawa, 2007; Das Evcimen & King, 2007). PKC pathway activation alters vasoconstriction and capillary permeability, and can cause hypoxia, angiogenesis, basement membrane thickening, and endothelial proliferation (Edwards, 1999; Williams, 1997). These changes in neurovascular blood flow are the likely source of PKC’s role in neuropathy, though further studies are needed to establish a strong connection. PKC activation also alters function of the Na+-K+ ATPase pump and other enzymes crucial to proper nerve conduction. Activation of different PKC isoforms has been shown to decrease Na+-K+ ATPase activity in smooth muscle cells and normalize activity in peripheral nerves (Greene, 1987; Xia, 1995). The link of PKC to diabetic neuropathy is supported by studies in streptozotocin (STZ) induced diabetic rats, where PKC inhibition normalizes both sciatic nerve blood flow and nerve conduction velocity (Nakamura, 1999). Overexpression of PKC isoforms can also directly induce insulin resistance (Cortright, 2000; Naruse, 2006).
4d. AGE Pathway
Non-enzymatic reactions between reducing sugars or oxaldehydes and proteins/lipids result in advanced glycation endproducts (AGEs) (Ahmed, 2005; Toth, 2007). Three main pathways are responsible for the formation of reactive dicarbonyls (AGE precursors): 1) oxidation of glucose to form glyoxal; 2) degradation of Amadori products (fructose-lysine adducts); and 3) aberrant metabolism of glycolytic intermediates to methylglyoxal.
AGEs are heterogeneous modified intracellular and extracellular biomolecules. Inside cells, both protein and DNA adducts alter function and cellular transport. Methylglyoxal, a highly reactive dicarbonyl, is shown to induce sensitivity to vascular damage in endothelial cells (Yao, 2007). Extracellular protein AGEs include plasma and matrix proteins that disrupt cellular adhesion and activate the receptor for AGEs (RAGE) (Ramasamy, 2007). AGE-RAGE interaction activates the transcription factor nuclear factor kappa B (NF-κB) (Figure 6). NF-κB regulates a number of activities including inflammation and apoptosis (Ramasamy, 2005). Activation of neuronal RAGE induces oxidative stress through NADPH oxidase activity (Vincent, 2007). Increased levels of AGE and RAGE are found in human diabetic tissue (Tanji, 2000). Diabetic RAGE knockout mice showed significant improvement in DPN and diminished expression of NFκB and PKC compared to wild type diabetic model (Toth, 2007). While the NFκB-PKC decrease in the knockout was present in dorsal root ganglion (DRG) and peripheral nerve, it was most pronounced in supporting Schwann cells. Collectively, the biochemical damage induced by AGEs results in impaired nerve blood flow and diminished neurotrophic support (Wada, 2005).
4e. PARP Pathway
PARP found in Schwann, endothelial cells, and sensory neurons is also implicated in glucotoxicity. PARP is a nuclear enzyme closely associated with oxidative-nitrosative stress: free radicals and oxidants stimulate PARP activation (Figure 6). Recent evidence also suggests that the two act in concert: PARP both causes and is activated by oxidative stress (Obrosova, 2005a). PARP acts by cleaving nicotinamide adenine dinucleotide (NAD+) to nicotinamide and ADP-ribose residues attached to nuclear proteins (Southan, 2003). The results of this process include NAD+ depletion, changes in gene transcription and expression, increased free radical and oxidant concentration, and diversion of glycolytic intermediates to other pathogenic pathways such as PKC and AGE formation (Du, 2003; Garcia Soriano, 2001; Ha, 2002; Obrosova, 2005a). Such PARP-implicated abnormalities manifest clinically as decreased nerve conduction velocity (NCV), small fiber neuropathy, neurovascular abnormalities, retinopathy, thermal and mechanical hyperalgesia, and tactile allodynia (Ilnytska, 2006; Li, 2005; Obrosova, 2005a; Obrosova, 2004; Pacher, 2002; Zheng, 2004).
4f. Oxidative Stress and Apoptosis
The AGE, polyol, hexosamine, PKC, and PARP pathways all contribute to neuronal damage. Figure 6 illustrates that the AGE and polyol pathways directly alter the redox capacity of the cell either through direct formation of ROS or by depletion of necessary components of glutathione recycling. The hexosamine, PKC, and PARP pathways exhibit damage through expression of inflammation proteins. The progression of diabetic neuropathy in a distal-proximal axon length-dependent manner suggests that damage is initiated in the axon (Leinninger, 2006b). Axons are susceptible to hyperglycemic damage both due to their direct access to nerve blood supply and their large population of mitochondria (Mt). Mounting evidence suggests that the hyperglycemic environment coupled with a compromised blood supply overloads the metabolic capacity of the Mt, producing oxidative stress (Brownlee, 2001). This oxidative stress leads to Mt damage followed by axonal degeneration and death.
Mitochondrial damage occurs due to excess formation of ROS and reactive nitrogen species (RNS) (Nishikawa, 2000; Obrosova, 2007; Obrosova, 2005c). ROS, such as superoxide and hydrogen peroxide, are produced under normal conditions through the Mt electron transport chain and are normally removed by cellular detoxification agents such as superoxide dismutase, catalase, and glutathione (Figure 7)(Leinninger, 2006b). Hyperglycemia leads to increased Mt activity, raising ROS production in the Mt. Peroxynitrite, the primary RNS, is formed by the reaction of superoxide and nitric oxide (NO). RNS induces a number of cytotoxic effects including protein nitrosylation and activation of PARP (Obrosova, 2005a; Obrosova, 2005b). Excessive ROS/RNS formation eventually overloads the natural antioxidant capacity of the cell, resulting in injuries to lipids, proteins, DNA. This damage ultimately compromises cellular function and integrity. As Mt are the origin of ROS/RNS generation, they are most susceptible to damage. While inhibitors of AR (Obrosova, 2002), PKC activation (The effect of ruboxistaurin on visual loss in patients with moderately severe to very severe nonproliferative diabetic retinopathy: initial results of the Protein Kinase C beta Inhibitor Diabetic Retinopathy Study (PKC-DRS) multicenter randomized clinical trial, 2005), AGE formation (Wada, 2001), and PARP (Ilnytska, 2006) can individually ameliorate hyperglycemia-induced nerve damage in animal models of diabetes, emerging evidence also suggests that these pathways converge to increase cellular oxidative stress (Figure 6). Cellular oxidative stress is further enhanced when excessive glucose leads to overproduction of superoxide as a byproduct of mitochondrial oxidative phosphorylation (Figure 7) (Vincent, 2004a). Overproduction of superoxide also markedly inhibits GAPDH, causing accumulation of upstream glycolytic intermediates. These intermediates further enhance AR, hexosamine, PKC, and AGE production, producing even more cellular injury. Experimental support for this unifying hypothesis derives from studies demonstrating that inhibition of superoxide accumulation by overexpression of superoxide dismutase prevents hyperglycemia-induced increases of AR (Nishikawa, 2000), hexosamine pathway products (Du, 2000), PKC activation, and AGE formation. Thus, there exists a vicious feed forward system in cells prone to diabetic complications, where glucose-activated metabolic pathways converge to produce cellular oxidative stress. Decreased nerve blood flow and ischemia, resulting from the processes described above, further exacerbate tissue injury. In summary, oxidative stress and ROS link the metabolic initiators and physiological mediators implicated in progressive nerve fiber dysfunction, damage, and loss in diabetic neuropathy. The generation of ROS may initiate a feed-forward cycle in which oxidative stress itself impairs anti-oxidative defense mechanisms.
Figure 7. Oxidative stress and mitochondrial dysfunction(Leinninger, 2006b).

Hyperglycemia increases production of reactive oxygen species (ROS) in mitochondria. NADH and FADH2 produced from the tricarboxylic acid cycle transfer to the mitochondria, where they serve as electron donors to the mitochondrial membrane-associated redox enzyme complexes. The electrons (e−) are shuttled through oxidoreductase complexes I, II, III and IV (cytochrome c), until they are donated to molecular oxygen, forming water. The electron transfer into complexes I, III and IV by NADH (and FADH2 via complex II to complex III) produces a proton gradient at the outer mitochondrial membrane, generating a potential between the inner mitochondrial membrane and outer mitochondrial membrane. This potential drives ATP synthesis, and is crucial for mitochondrial viability, function, and normal metabolism. As electrons are passed from complex II to complex III, however, ROS are produced as by-products. The levels of ROS produced during normal oxidative phosphorylation are minimal, and they are detoxified by cellular antioxidants such as glutathione, catalase and superoxide dismutase. The hyperglycemic cell, on the other hand, shuttles more glucose through the glycolytic and tricarboxylic acid cycles, providing the cell with an over-abundance of NADH and FADH2 electron donors. This produces a high proton gradient across the inner mitochondrial membrane, which increases the turnover of the initial complexes, and thereby produces increased levels of radicals. Accumulation of these radicals, or ROS, is severely detrimental to mitochondrial DNA, mitochondrial membranes and the whole cell. Abbreviations: Cyto-c, cytochrome c; CoQ10, coenzyme Q10; e−, electrons; GSH, glutathione; GSSG, oxidized glutathione; H2O2, hydrogen peroxide; O2•−, superoxide; Pi, phosphate; SOD, superoxide dismutase.
In addition to their role in metabolism, Mt are involved in the determination of cell fate and viability. Oxidative stress not only damages Mt DNA, proteins, and membranes, it also initiates signaling pathways that result in localized mitochondrial destruction called mitoptosis. One pathway essential to mitoptosis, and subsequently apoptosis, involves Mt division via the dyanmin related protein 1 (Drp1) (Frank, 2001; Lee, 2004b). Mt normally undergo an equilibrium-driven process of fission and fusion. In times of stress, Drp1 translocates from the cytosol to the Mt so as to increase Mt fission events (Arnoult, 2005). Aberrant Mt fission is associated with mitoptosis and implicated in apoptosis. Increased levels of Drp1 are found in vitro and in vivo models of diabetic neuropathy (Leinninger, 2006a). This implicates Mt fission in diabetic neuropathy and renders Drp1 a potential therapeutic target.
Mt are pivotal components of metabolism, oxidative stress, and programmed cell death/apoptosis (Russell, 1999). As such, neurons in a hyperglycemic environment display signs of both oxidative stress and apoptosis (Russell, 1999). Diabetic models have shown either apoptosis in the cell body or neuroaxonal dystrophy. Some studies have shown an absence of apoptosis in high glucose treated sensory neurons (Cheng, 2003; Gumy, 2008). The current hypothesis that counts for these observations is that in vivo, recurrent injury occurs, activating cell death pathways; initially neurons with support from glia are able to undergo successful repair, however eventually cellular defense is overcome by recurrent activation of injury pathways, causing damage to the cell body. Over time, this cycle injures Mt, alters Mt distribution, and initiates axons dying back toward the cell body (Sullivan, 2005; Vincent, 2004b).
4g. Inflammation
Inflammatory agents including C-reactive protein and TNF-α are present in the blood of both T1DM and T2DM (Gomes, 2003; Gonzalez-Clemente, 2005). Higher levels of these proteins correlate with the incidence of neuropathy (Gonzalez-Clemente, 2005). Recent data from the Eurodiab prospective complications study demonstrates a correlation between diabetic neuropathy and plasma levels of hsp 27 (Gruden, 2008). Hsp 27 is a required intermediate in the pathway of TNF-α induction of the inflammatory mediators cyclooxygenase-2 (Cox-2), IL-6, and IL-8. The production of the initiating inflammatory mediators TNF-α and TGF-β results from several of the glucose-induced pathways already outlined (Brownlee, 2005; Vincent & Feldman, 2004a). As illustrated in Fig. 4, when excess glucose is shunted through alternative metabolic pathways such as the fructose-6-phosphate or diacylglycerol, the signaling intermediates and modified transcription factors lead to increases in TGFβ and NFκB (Brownlee, 2001). Similarly, breakdown of glycolytic triose phosphates forms methylglyoxal, an AGE, that covalently modifies transcription factors (Yao, 2007). One specific consequence of these modifications is decreased binding of a repressor of angiotensin II, known as Sp3. Thus, antiotensin II increases and leads to activation of vascular endothelial cells (Yao, 2007). In the endoneurium, this activation leads to inflammatory cell recruitment, local generation of cytokines, and reduced blood flow that leads to further generation of ROS (Coppey, 2006). Other extracellular AGEs that activate RAGE also lead to intracellular inflammatory signaling to upregulate NF-κB (Toth, 2007).
Cox-2 is an important enzyme that is unpregulated by NF-κB (Lee, 2004a). This upregulation is observed in peripheral nerves and vascular tissues in experimental diabetes (Kellogg, 2005). Cox-2 activity appears to drive a feedforward loop since Cox-2 is upregulated by NF-κB and in turn it generates prostaglandin E2 and ROS that activate NF-κB. Pharmacological blockade or gene ablation of Cox-2 prevents diabetes-induced changes in peripheral nerves including depletion of GSH, increases in TNF-α, and blood flow and nerve conduction deficits (Kellogg, 2007; Matsunaga, 2007).
Another inflammatory enzyme regulated by NF-κB is inducible nitric oxide synthase (iNOS) (Kim, 2008). Like Cox-2, iNOS both induces and is induced by NF-κB, leading to a vicious cycle of inflammation (Hasnis, 2007; Kim, 2008). The NO generated by iNOS directly modulates the blood supply to nerves and participates in microvascular changes following injury (Levy, 2004; Zochodne, 2005). NO has direct roles in axon and myelin breakdown following an injury and also contributes to the development of neuropathic pain (Levy & Zochodne, 2004; McDonald, 2007). Excessive local levels of NO during inflammation may damage axons and growth cones (Zochodne & Levy, 2005).
All of the inflammatory mechanisms in diabetic neuropathy appear to converge upon the activation of NF-κB. Because of chronic NF-κB activation, blood vessels and nerve cells are more susceptible to injury in ischemia reperfusion (Wang, 2006). Subsequent to ischemia-reperfusion there is extensive infiltration of monocyte macrophages and modest infiltration of granulocytes in diabetic peripheral nerves (Wang, 2006). The cytokines induced by NF-κB in endothelial cells, Schwann cells and neurons also lead to macrophage recruitment in diabetic nerves (Yamagishi, 2008). Macrophages promote diabetic neuropathy through a variety of mechanisms, including production of ROS, cytokines and proteases, which result in myelin breakdown and cellular oxidative damage (Conti, 2002; Kawamura, 2008; Tesch, 2007). Excessive macrophage recruitment likely impairs nerve regeneration in diabetic neuropathy (Conti, 2002; McDonald, 2007).
4h. Growth Factors
Growth factors promote the growth and survival of neurons and direct neurite outgrowth (Leinninger, 2004). Given that diabetic neuropathy is characterized by neuronal degeneration and damage to supporting Schwann cells, perturbations in growth factors such as nerve growth factor (NGF), insulin like growth factor (IGF), and neurotrophin 3 (NT3) have been suggested to be involved in the pathogenesis of diabetic neuropathy. These factors bind to heterodimeric tyrosine kinase receptors. The receptors for the NGF family of growth factors consist of the p75NTR and a specific trk tyrosine kinase, which confers ligand specificity (although there is some overlap).
Expression levels of multiple growth factors are altered in animal models of DPN. NGF is the most studied growth factor in diabetic neuropathy. NGF is produced by muscle and keratinocytes, and its trkA receptor is expressed on sensory and sympathetic neurons (Averill, 1995; Fang, 2005; McMahon, 1994; McMahon, 1995). In multiple diabetic models, NGF levels are reduced as well as retrograde transport of NGF diminished (Hellweg, 1994; Kasayama, 1989). Interestingly, when glucose levels are returned to normal levels, NGF levels return to normal. This indicates that diabetes, either due to hyperglycemia or by lack of insulin, has the capacity to regulate growth factors (Hellweg, 1991). Some studies have generated conflicting results with regard to NGF expression levels (Arrieta, 2005; Delcroix, 1998; Delcroix, 1997; Fernyhough, 1994, 1995a; Fernyhough, 1995b; Hellweg, 1990; Hounsom, 1998). Despite these discrepancies, an observed decrease in the retrograde transport of NGF (both endogenous and exogenous) in diabetic rats is noteworthy in that the transport of NGF to the soma is required for its neurotrophic effects to occur (Arrieta, 2005; Fernyhough, 1995b; Hellweg, 1994; Schmidt, 1983; Schmidt, 1985). Similar to NGF, IGF I & II are down-regulated under diabetic conditions, though administration of insulin restored expression (Migdalis, 1995; Olchovsky, 1991; Wuarin, 1994). NT-3 is expressed in muscle and skin. It can signal through trkA and B to some extent, and primarily signals through trkC, suggesting broad therapeutic potential (Barbacid, 1994; Lewis, 2006; Lindsay, 1996). Like trkB, trkC is found in motor neurons, and a population of large-diameter sensory neurons responsible for proprioception and tactile sensation (Barbacid, 1994; Lindsay, 1996). As with studies of other growth factors, changes in NT-3 expression in diabetes have not been consistently documented. NT-3 proteins levels are upregulated in diabetic sural nerve, though mRNA levels have been reported as both increased and decreased (Cai, 1999; Rodriguez-Pena, 1995).
5. Therapeutic Strategies for Diabetic Neuropathy
5a. Glycemic Control
Therapies for DPN and DAN may be divided into treatments that target the underlying pathogenetic mechanisms (Boulton, 2004a; Singh, 2005; Trotta, 2004) and those aiming to relieve symptoms (Adriaensen, 2005). In the latter category there are numerous established approaches; in the former, the only proven method currently available to prevent DPN and DAN or slow progression is strict glycemic control (Tesfaye, 2005).
5ai. T1DM
DCCT
As discussed previously, the DCCT compared intensive treatment (3 or more insulin injections or insulin pump) aiming to normalize HbA1c with conventional treatment (1 or 2 insulin injections aiming to prevent hyperglycemic symptoms and hypoglycemia) in 1,441 patients with T1DM. After 5 years of follow-up, the prevalence of confirmed clinical neuropathy (defined by history or physical exam and confirmed by either abnormal nerve conduction or ≥ 1 autonomic function test) was 64% lower in patients receiving intensive treatment (HbA1c = 7.2%) than in those receiving conventional treatment (HbA1c = 9.1%). Further, nerve conduction velocities remained stable in patients receiving intensive treatment and declined significantly in those assigned conventional treatment (The effect of intensive diabetes therapy on the development and progression of neuropathy. The Diabetes Control and Complications Trial Research Group, 1995). The results of the DCCT agree with a similar study in Europe, the EURODIAB trial (Tesfaye, 1996).
It has been reported that the benefits of tight glycemic control in reducing microvascular complications are more far-reaching than originally determined. The Epidemiology of Diabetes Interventions and Complications (EDIC) study has followed the DCCT cohort for more than 7 years after the termination of the original study. EDIC has shown that despite convergence of mean HbA1c in patients originally randomized to intensive vs. conventional treatment (due to institution of intensive treatment in 75% of the former conventional treatment group and a small proportion of the original intensive treatment group choosing to relax glycemic control), the rate of progression of retinopathy and nephropathy in patients originally randomized to intensive treatment remained significantly below those in the original DCCT conventional treatment arm (Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus, 2002). This benefit was also reported for neuropathy. At the end of EDIC Year 1, mean HbA1c levels were 7.9% and 8.3% in the former intensive and conventional DCCT arms, respectively. By EDIC Year 5 the HbA1c levels were statistically indistinguishable (8.1 vs. 8.2%, P = 0.09). However, there continues to be a slower rate of acquisition and progression of DPN in the patients from the former intensive treatment arm, despite over 8 years of similar control(Martin, 2006).
Continuous Glucose Monitoring
Despite the evidence of the DCCT which suggest that HbA1C levels are strong predictors for diabetic complications, factors other than average blood glucose levels have a profound influence on incidence of diabetic complications. Close examination of the DCCT has indicated that the prognosis of complications may need to go beyond the average blood glucose level as indicated by HbA1C (Brownlee, 2006). Figure 8 shows that when the conventional therapy group of the DCCT was examined, the HbA1C shows a general correlation with incidence of diabetic complications (Hirsch, 2005). In contrast, when the intensive therapy group was examined, HbA1C holds only a slight relation to development of diabetic complications. As the difference between intense therapy and conventional therapy was multiple insulin injections and blood glucose monitoring, intensive therapy cohort patients are considered much less likely to undergo dynamic glycolytic flux. These studies suggest that glycemic variability rather than average glycemic control (HbA1C) would be a better target for diminishing the onset and progression of complications.
Figure 8. Risk of Diabetic Complications (Retinopathy) in Control vs. Intensive Therapy as related to HbA1C.
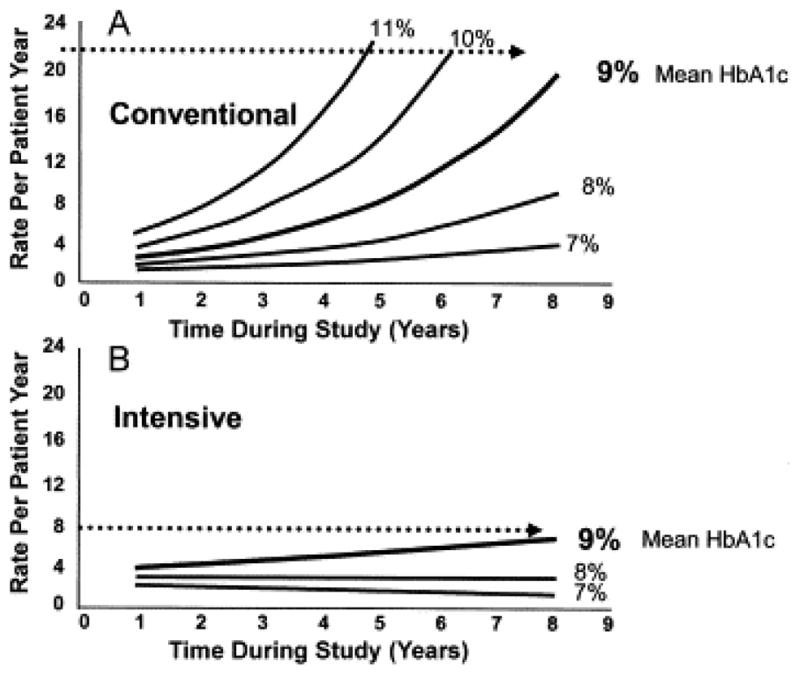
Absolute risk of sustained retinopathy progression as a function of updated mean A1C (percentage) during the DCCT and the time of follow-up during the study (years), estimated from absolute (Poisson) regression models. (A) Conventional treatment group. (B) Intensive treatment group. Results suggest that average glucose levels may be less important to prognosis of complications than fluctuations in glucose levels. Reprinted with permission from DCCT Research Group (1995) (Hirsch & Brownlee, 2005)
In vitro studies now show that hyperglycemia-induced superoxide resulting in oxidative stress is the major causal agent of cellular injury. Work by Monnier et al. showed no correlation between oxidative stress and 24 h mean glucose concentration, fasting plasma glucose levels or HbA1C (Monnier, 2006). On the other hand there was a direct correlation between oxidative stress and glycemic variability (as determined by mean amplitude of glycemic excursion). These data indicate that glycemic flux and variability lead to oxidative stress.
Glycemic variability is directly correlated to oxidative stress and is likely a better predictor of diabetes complications. Thus, the ability to reduce glycemic variability holds the potential of preventing complications. Until recently, real-time tracking and correction of glycemic flux was unattainable. Continuous glucose monitors (CGM) are now available which measure blood glucose levels every 1–5 min. Glucose self-monitoring is recommended 3 or more times per day. With the implantable CGM, >120 glucose levels per day are registered and stored (FreeStyle NavigatorTM Continuous Glucose Monitoring System Use in Children with Type 1 Diabetes using glargine-based multiple daily dose regimens: Results of a Pilot Trial, 2007;, Relative accuracy of the BD Logic and FreeStyle blood glucose meters, 2007; Wilson, 2005). Real time data from the CGM allows for the patient to adjust insulin or food intake before either hyper- or hypo-glycemia occurs. As such, glycemic variability may be reduced by modulating therapy (insulin/glucose intake) in response to glucose trends. Implantable CGM sensors are used for between 3 and 7 days. Widespread use of the CGM remains limited at present, likely due to the cost to the patient.
Islet Transplantation
Pancreas transplantations in T1DM patients are now at a sufficient success rate to assess potential therapeutic effects on diabetic complications. Pancreas/islet transplantation re-established normoglycemia in T1DM patients. Compared to T1DM patients who received only kidney transplants, T1DM patients with successful pancreas transplants (as determined by normoglycemia and insulin independence) showed improvements in motor and sensory nerve conduction and clinical neuropathy (Navarro, 1997). These improvements were seen after 3.5 years for clinical examinations, between 1–5 yrs for sensory nerve conduction and after 10 years for motor nerve conduction. In fact, 50% of islet transplant patients with DPN exhibited stabilization or improvement of their neuropathy (Lee, 2005). Improvements were seen only for DPN and not for DAN.
5a ii. T2DM
Evidence that good glycemic control can delay or prevent progression of diabetic neuropathy in patients with T2DM is more limited. Neuropathy findings from the UKPDS were discussed previously. In brief, although it was clearly shown that intensive treatment significantly reduced the risk of an aggregate endpoint of microvascular complications, and single endpoints of retinal photocoagulation, and cataract extraction, there was only a trend for reduction in the risk of amputation (P = 0.099) in the intensively-treated patients (Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group, 1998). There was, however, a significant reduction in the risk of sudden death (P = 0.047) in which it is possible that autonomic neuropathy plays a role.
In the feasibility study for the Veterans Administration Cooperative Study on T2DM (VA CSDM), the effects of 2-year intensive treatment on clinical DPN and on DAN was examined in 153 men with T2DM (average duration = 7.8 ± 4 yrs) (Azad, 1999). Intensive treatment (stepwise introduction of multiple insulin injections plus daytime glipizide) achieved a separation in HbA1c between intensive and conventional treatment of 2.1%, with intensively-treated patients at or below 7.3%. The baseline prevalence of clinical DPN was ~ 50% and of abnormal autonomic function tests, was ~ 35%. The prevalence of clinical DPN and abnormal autonomic function tests increased significantly and similarly in the two treatment groups, as did the prevalence of erectile dysfunction. Although there was a nearly-significant decrease in the prevalence of cranial neuropathy and more frequent preservation of touch sensation in the upper extremities in the intensive vs. conventional treatment group, this study provided only minimal evidence that good glycemic control improves neuropathic outcomes in patients with T2DM (Azad, 1999).
In summary, the interventional evidence relating hyperglycemia and neuropathy in patients with T2DM is less overwhelming than that in patients with T1DM. Yet, in view of the strong association between HbA1c and the incidence, prevalence, and progression of neuropathy in all forms of diabetes, aggressive treatment aiming for normalization of both fasting and postprandial glucose levels remains the first and most important step in treating patients with any form of diabetic neuropathy.
There is an uncommon form of neuropathy that may occur in poorly-controlled diabetic patients shortly after instituting aggressive insulin therapy. This acute painful neuropathy has also been described in other situations, particularly in male patients following rapid weight loss (Windebank, 2001). This differs from DPN in that sensory loss is minimal and weakness does not occur. In the cases arising from rapid weight loss, improving glycemic control and recovery of weight lead to symptomatic improvement, and temporarily relaxing glycemic control leads to symptomatic relief in those cases appearing to arise from instituting aggressive insulin therapy. In this form of neuropathy, complete resolution usually occurs over 6 to 24 months.
Other than strict glycemic control, disease modifying treatments for neuropathy are presently only experimental. Some have progressed to clinical trials and others, based on new findings from work on mechanisms of neuropathy, such as the central role of reactive oxygen species, are in earlier stages of development. These will be discussed at the end of this review.
Symptomatic Treatment of Peripheral Neuropathy. Foot Care
All patients with diabetes should receive at least an annual foot examination to identify high-risk conditions (Association, 2004; Boulton, 2005). Since DPN is one of the most important predictors of foot ulcers and amputation, the importance of preventative foot care in neuropathic patients cannot be overemphasized. Neuropathic patients should undergo a careful foot examination at every office visit and promptly be referred to a foot care specialist whenever necessary. Patient education on proper foot care including shoe selection, nail trimming, and daily foot inspection is essential, and patients with sensory loss should be advised to inspect their shoes three times daily to be sure that no sharp objects are present. Any signs of local infection should be treated by medical and surgical means if necessary (Boulton, 2004a; Singh, 2005).
5b. Symptomatic Therapies
5b1. Diabetic Polyneuropathy
Pain is an early manifestation of diabetic neuropathy and frequently precedes the diagnosis of diabetes (Boulton, 2005; Feldman, 2005). Several recent studies suggest that nearly one third of the patients with impaired glucose tolerance (pre-diabetes) seek medical attention for a pain syndrome identical to DPN (Singleton, 2005; Singleton, 2003). While DPN is a persistent symptom in epidemiological studies of patients with T2DM, it is less common in type 1 diabetes(Barrett, 2007; Clarke, 2002; Currie, 2007). Clinically, DPN includes burning, tingling, electric-like, achy pain beginning in the feet and extending upward over time. Patients with DPN also experience allodynia and hyperalgesia. Allodynia occurs when normally nonpainful stimuli become painful, whereas hyperalgesia is increased sensitivity to normally painful stimuli. DPN is a major factor in decreased quality of life for patients with diabetes(Dworkin, 2007a; Dworkin, 2005; Jensen, 2006a). While estimates vary, approximately half of all patients with diabetic neuropathy of T2DM experience pain, usually at the onset of their disease. Patients with DPN develop insomnia, depression and anxiety, decreased mobility, psychomotor impairment and inability to work(Dworkin, 2007a; Dworkin, 2007b; Dworkin, 2005; Jensen, 2006a; Jensen, 2006b). Over a period of time that could last several years, DPN subsides and the disabling pain is replaced by a complete loss of sensation, leading to the numb, insensate diabetic foot(Feldman E.L., 1999; Feldman, 2005; Feldman, 2002b).
Our lack of understanding of the pathogenesis of this disorder precludes the development of mechanism-specific therapies (Feldman, 2005; Feldman, 2002a). Therefore, currently accepted medical approaches are only partially successful and are often ineffective (Dworkin, 2007a; Dworkin, 2005). These include the use of anti-convulsants, anti-depressants, topical agents, and opioid based therapies (Ziegler, 2006) that all have undergone placebo-controlled studies in patients with DPN (Table 10).
Table 10.
Therapeutics for the Polyol Pathway
| Compound | Trials/Notes | References |
|---|---|---|
| Sorbinil | Only slight improvement in NCV; high rate of skin rash, trial withdrawn | (Judzewitsch, 1983) |
| Tolrestat | Halted mild diabetic neuropathy progression; no significant improvements in NCV, trial withdrawn | (Boulton, 1990; Giugliano, 1993) |
| Ponalrestat | No effect due to poor pharmacokinetics & pharmacodynamics, trial withdrawn | (Greene & Sima, 1993; Stribling, 1985) |
| Zopolrestat | Low levels: slight NCV improvement High levels: significant improvement in NCV; elevated liver enzymes, trial withdrawn |
(Arezzo, 1996) |
| Zenarestat | Dose dependent improvement of NCV | (Gabbay, 2004; Greene, 1999) |
| Lidorestat | Withdrawn at Phase 2 clinical trial | |
| Fidarestat | Similar to Sorbinil, suspended in phase 3 due to resource consolidation | (Giannoukakis, 2003) |
| AS-3201/ Ranirestat | Promising Phase 2 trials; Phase 3 underway. High placebo effect complicating study | (Bril & Buchanan, 2006; Oates, 2008, in press) |
| Epalrestat | Delayed progression of diabetic neuropathy, study not replicated | (Hotta, 2006) |
| Myo-inositol | Animal studies indicate beneficial effects, human studies needed | (Sima, 1997) |
NCV=Nerve Conduction Velocity
Antidepressants
Tricylic and tetracyclic reagents
The tricylic and tetracylic antidepressants (TCA) are considered as the first line treatment for neuropathic pain. These antidepressants control pain and pain related symptoms such as insomnia and depression. The therapeutic actions of these agents are mediated by inhibition of the reuptake of norepinephrine and serotonin. In a study reported by Max et al., amitriptyline (150 mg/day) is superior to placebo in relieving DPN after 6 weeks of treatment (Max, 1992). However, amitriptyline is associated with significant side effects, including dry mouth, sedation, and blurred vision. Desipramine is better tolerated at 111 mg/day and is as effective as amitriptyline in alleviating DPN (Max, 1992). Randomized control trials for imipramine demonstrated that a dose of 50 and 75 mg/day significantly improves DPN (Sindrup, 1989; Sindrup, 1990b; Sindrup, 1999b). In addition, clomipramine relieves the symptoms of DPN (Sindrup, 1990c). Pooling the data from all of these trials suggests that approximately 1 in 3 patients experience at least 50% relief from pain by using these drugs (Collins, 2000). The use of TCA is limited by their side effects (Jann, 2007). Overall, secondary amines (nortriptyline, desipramine) are better tolerated than tertiary amines (amytriptyline, imipramine) (Dworkin, 2007b). TCAs are not well tolerated in older patients. The TCAs should be used with great caution (or avoided altogether) in patients with cardiac arrhythmias, congestive heart failure, orthostatic hypotension, urinary retention, or angle-closure glaucoma (Simmons, 2002). It is important to note that TCAs are contraindicated in patients taking monoamine oxidase (MAO) inhibitors. The usual dosage schedule for TCAs is 10 to 25 mg at bedtime initially, titrating as tolerated up to 100 or 150 mg as a single bedtime dose. In addition, their analgesic effects require several weeks to develop which limits their utility for acute pain (Max, 1987).
Selective Serotonin Reuptake Inhibitors and Serotonin-Norepinephrine Reuptake Inhibitors
The selective serotonin reuptake inhibitors (SSRIs) are newer antidepressants that have largely replaced TCAs for the treatment of depression because they are better tolerated. However, in contrast to TCAs, the effects of SSRIs are limited in DPN. Fluoxetine, 40 mg/day, is not different from placebo (Max, 1992). In a crossover study with paroxetine and imipramine, significant benefits from paroxetine 40 mg/day are observed (Sindrup, 1990b). The improvement is less than imipramine 50 mg/day but better than placebo. Citalopram 40 mg/day has also been shown to be better than placebo for treating DPN (Sindrup, 1992). However, the numbers of patients involved in most of these studies are small and the trial periods are all short, limiting the interpretation of these data. Pooled data for SSRI treatment of DPN demonstrates no significant difference between SSRIs and placebo (Collins, 2000). In contrast, Tramadol is a weak μ-receptor agonist that inhibits re-uptake of serotonin. A double blind, randomized, placebo controlled trial using an average dose of 210 mg/day for 6 weeks produced significantly reduced pain scores in patients with DPN (Harati, 1998). Nausea, constipation, headache, and dyspepsia were common side effects. Another trial also demonstrated tramadol 200–400 mg/day significantly relieves DPN over placebo (Sindrup, 1999a). Tramadol is well tolerated with only mild side effects. In addition, a combination of tramadol/acetaminophen (37.5/325 mg) when taken as 1–2 tablets four times a day is effective in alleviating DPN (Freeman, 2007).
The serotonin-norepinephrine reuptake inhibitors (SNRI) have greater efficacy against DPN than SSRIs. Duloxetine has been approved by the Food and Drug Administration (FDA) for treating DPN following three large randomized placebo control trials (Goldstein, 2005; Raskin, 2005; Wernicke, 2006). In these trials, duloxetine 60 mg and 120 mg daily provided significant relief from DPN. The higher dose provides greater relief from DPN but is associated with increased side effects. In general, duloxetine is better tolerated, in terms of gastrointestinal and cardiac side effects, than other serotonin-norepinephrine reuptake inhibitors. Venlafaxine 150–225 mg/day alleviates DPN but produces unacceptable cardiac side effects with increased risk of electrocardiographic changes (Rowbotham, 2004).
Anticonvulsants
Anticonvulsants control neuronal excitability by blocking sodium and/or calcium channels (Wiffen, 2005). Originally developed for preventing seizures, they are in broad use for the treatment of neuropathic pain. Phenytoin and carbamazepine primarily block the voltage gated sodium channel. At doses between 200–600 mg/day, both reduce DPN compared to placebo. Due to side effects and newer improved therapies, these compounds are not recommended (Chadda, 1978; Gomez-Perez, 1996; Rull, 1969; Saudek, 1977).
Sodium valproate enhances GABA levels in the central nervous system, inhibits T type calcium channels, and increases potassium inward currents. Again, side effects, including hair loss, weight again, hepatotoxicity, and cognitive dysfunction are not insignificant and increase with long-term use, although a dose of 500 mg/day decreases DPN (Kochar, 2004).
Lamotrigine is a new anticonvulsant which blocks voltage gated sodium channels, decreases presynaptic calcium currents to inhibit the release of glutamate, and increases GABA levels in the brain. Eisenberg and colleagues reported favorable results of lamotrigine (≤ 400 mg/day) against DPN (Eisenberg, 2001). Vinik et al. also reported two large scale (n=360) randomized double-blind, placebo-controlled trials. Although there is a reduction on the pain scale in one trial after 19 wk of duration with lamotrigine (400 mg/day), there is no difference between lamotrigine and placebo group at the end of the trial (Vinik, 2007). Lamotrigine is well tolerated but its efficacy against DPN is questionable.
Topiramate has multiple actions: 1) blocking activity-dependent voltage gated sodium channels; 2) inhibiting L-type voltage gated calcium channels; and3) blocking postsynaptic kainite/α-amino-3-hydorxy-5-methyl-4-isoxazolepropionic acid (AMPA) excitatory amino acid receptors. Raskin et al reported a randomized, double-blind, placebo-controlled study which involved 323 patients with DPN (Raskin, 2004). Topiramate ≤ 400 mg /day was usually well tolerated and significantly alleviated DPN in approximately 1 out of 6 patients.
Zonisamide blocks both voltage dependent sodium channels and T-type calcium channels. A randomized, double blind, placebo-controlled pilot study of 25 patients with a mean dose of 540 mg/day did not significantly reduce DPN after 6 weeks of titration and maintenance treatment (Atli, 2005). Common side effects were restlessness, GI discomfort, headache, and weight loss.
Oxcarbazepine is a keto-analogue of carbamazepine, which blocks sodium channels. In one study, ≤1800 mg/day oxcarbazepine significantly reduced DPN (Dogra, 2005), but in a larger study, no significant reduction of DPN was seen with 1200 and 1800mg/ day (Beydoun, 2006). Oxcarbazepine has a good side effect profile and is well tolerated. However, more studies are necessary to clarify its potential for treating DPN.
Calcium Channel α2-δ ligands
Gabapentin is widely used for neuropathic pain due to its effectiveness and relatively fewer side effects than TCA and other anti-convulsants. Gabapentin produces analgesia via binding to the α2-δ site of L-type voltage gated calcium channels and decreasing calcium influx. Gabapentin ≤ 2400 mg/day is effective in treating DPN compared to amitryptyline (≤ 90 mg/day) according to a randomized control trial of 165 patients (Backonja, 1998; Dallocchio, 2000). However, another study found no difference between gabapentin (900–1800 mg/day) and amitriptyline (25–75 mg/day) (Morello, 1999). Large head-to-head comparison studies are needed to demonstrate the superiority of gabapentin over amitriptyline. Gabapentin is usually well tolerated with slow titration. Common side effects of gabapentin include dizziness, ataxia, sedation, euphoria, ankle edema, and weight gain. Moreover, it usually takes weeks of titration to reach the maximal effective dose (Dworkin, 2007b) and a dosing of 3 per day is often necessary.
Like gabapentin, pregabalin also acts by binding to the α2-δ subunit of calcium channels. As demonstrated in four randomized placebo control trials, pregabalin (300–600 mg/day) is significantly more effective in alleviating DPN than placebo (Freynhagen, 2005; Lesser, 2004; Richter, 2005; Rosenstock, 2004). Unlike gabapentin, pregabalin has better GI absorption and can be administered twice per day. Its linear pharmacokinetics provide a rapid (< two weeks) onset of maximal pain relief (Dworkin, 2007b). However, side effects are similar to Gabapentin with dizziness, ataxia, sedation, euphoria, ankle edema, and weight gain. Among these side effects, weight gain is especially concerning for patients with T2DM. Like duloxetine, pregabalin is approved by FDA for treating DPN.
Mexelitine
Mexelitine is an anti-arrythmia medication and has been used for treating a variety of painful neuropathic conditions including DPN (Jarvis, 1998). Several randomized placebo control trials have been performed, but none of the studies revealed greater than 50% reduction in pain scores. However, those patients with stabbing or burning pain, heat sensations, or formication benefit most by mexiletine therapy (Stracke, 1992).
Opiates
Slow release oxycodon 20 mg/day relieves DPN over a 6-week period (Gimbel, 2003). In a crossover design treatment strategy, slow release oxycodon was effective against DPN at a maximum dose of 80 mg/day (Watson, 2003). Although opioids are effective against DPN, long term use of opioids will result in side effects including constipation, urinary retention, impaired cognitive function, impaired immune function, and many other issues associated with tolerance and addiction. Recenly, trials used combination of therapies with opioid and Gabapentin has proven there is an additive effect of pain relieve in comparison to individual treatment. Gilron et al tested patients with neuropathic pain from DPN and postherpetic neuralgia using maximal tolerated doses of morphine, Gabapentin or both(Gilron, 2005a). The combinations of drugs achieve higher potency of pain relieve than individual treatment. However, the maximal tolerated dose of each drug is lower in the patient group with combination therapy, suggesting increased side effects with drug combination. The combination pharmatherapy have been used widely in clinical practice but more studies need to be performed to establish safety, compliance and cost-effectiveness and determine optimal drug combinations and dose ratios; comparing concurrent with sequential combination therapy; and combining more than two drugs(Gilron, 2005b).
Non-steroidal anti-inflammatory drugs
Non-steroidal anti-inflammatory drugs (NSAID) are a class of medications that inhibits cyclooxygenases, and thus prevent the formation of prostaglandins. Usually, NSAID are not recommended for the treatment of DPN due to their detrimental effects to GI, renal, and cardiac functions. Risk of overdose is also high in patients with chronic pain. However, a small, single blinded study demonstrated ibuprofen 2400 mg/day and sulindac 400 mg/day significantly reduced the paresthesia scores of DPN at 24 weeks (Cohen, 1987).
N-methyl-d-aspartate receptor antagonists
Two N-methyl-d-aspartate (NMDA) receptor antagonists, dextromethorphan and memantine, have been tested in DPN (Sang, 2002). These placebo controlled cross over studies involve 23 patients with diabetic neuropathy. The studies were designed with a 7-week titration period followed by a 2-week maintenance period for lorazepam, an active placebo, or one of the NMDA inhibitors. Both high and low doses of the inhibitors were assessed. Treatment with dextromethorphan but not memantine produced a significant dose-dependent decrease in DPN. However, the NMDA inhibitors have significant side effects, including sedation, dry mouth, and gastrointestinal distress.
Topical agents
Capsaicin is an extract of capsicum peppers. Capsaicin binds to TRPV1 receptor and exhausts substance P in the peripheral nerves to achieve it analgesic effects. In the study published by the Capsaicin Study Group, 0.075% capsaicin cream applied three times a day for 6 weeks was more effective in alleviating DPN than placebo (The Capsaicin Study, 1992). Burning was the most common side effects which tended to decrease as therapy is continued. The therapeutic effects of capsaicin started weeks after the cream application. Recently, a patch containing high concentrated capsaicin has demonstrated promising effects in treating diabetic pain.
Because impaired NO generation leading to reduced blood flow may be involved in DPN, a small trial using isosorbide dinitrate, an NO donor, was performed. In a 12 week, double blind, placebo controlled, crossover study with 22 patients, isosorbide dinitrate spray significantly relieved DPN (Yuen, 2002). Patients in the trial reported minor headaches and a larger sized study is necessary to evaluate the potential use of this treatment for DPN.
Topical lidocaine 5% patches have been reported by several studies to relieve DPN. In an open labeled study, up to four 5% lidocaine patches applied for up to 18 h/day are well tolerated in patients with painful diabetic polyneuropathy. Lidocaine patches significantly improved pain and quality-of-life ratings, and may allow tapering of concomitant analgesic therapy (Barbano, 2004). Given the open-label design of this trial, a randomized controlled trial is necessary to confirm these results.
Selection of a Drug Regimen for DPN
Recently, the data from most available placebo-controlled trials was used to generate an algorithm and a flow chart (Figure 9) for treating neuropathic pain (Dworkin, 2007b; Strokov, 2000). The consensual first line regimens are TCA, calcium channel α2-δ ligands, topical lidocaine, and SNRIs. The recommended second line treatments are tramadol and opioids. The third line treatments include anti-epileptics, other antidepressants (SSRIs), NMDA receptor blockers, mexiletine, and topical capsaicin.
Figure 9. Management of Neuropathic Pain.

Stepwise instructions for treatment of the symptoms for painful diabetic neuropathy based on multiple levels of medications, pain scores and evaluations. Adapted from (Dworkin, 2007b)
Non-drug treatments that have shown efficacy in controlling pain associated with DPN include transcutaneous electrical nerve stimulation (TENS) (Kumar, 1997) and acupuncture (Abuaisha, 1998). There is, however, a notable absence of controlled clinical trials of these treatments. Unfortunately for patients refractory to the stepwise approach to drug treatment as outlined above, the prognosis is poor with many patients requiring long-term care in a pain clinic.
Placebo Effect
Recent work by Tesfaye et al. has elucidated an interesting longlasting placebo phenomenon in DPN(Tesfaye, 2007). Two 1 yr placebo controlled DPN studies of ruboxistaurin were analyzed. After studying the DPN, all of which were characterized as mild for inclusion, progressive improvenments in symptoms were demonstrated, whereas nerve conduction studies and heart rate deep breating worsened over the course of 1 yr. This studying indicates a need to critically develop the timeframe and endpoints of clinical studies for DPN. To establish clinically significant worsening of DPN will require timeframes greater than 1 yr.
5b ii. Diabetic Autonomic Neuropathy
As discussed previously, the first step in the treatment of all forms of diabetic neuropathy is long-term improvement of glycemic control. However, there is little evidence to suggest that even strict glycemic control can reverse DAN, although it is necessary to prevent progression. Since the symptoms of DAN may be manifested in essentially any organ, symptomatic treatment is obviously aimed at the affected organ or body system. It should be reemphasized that a careful history and knowledge of all prescription and non-prescription medications is essential to rule out other causes other than diabetic autonomic neuropathy and identify any concomitant condition(s).
Orthostatic hypotension is notoriously difficult to manage because the standing blood pressure must be raised without causing hypertension when the patient lies down. Options for the treatment of orthostatic hypotension are listed in Table 7. Non-pharmacologic treatment should be the initial approach. To increase venous return, supportive stockings should be worn during the day and removed at bedtime. Patients should be advised to avoid hot baths, to get out of bed or stand up slowly and if their diabetes is being treated with insulin, patients should administer insulin injections while lying down.
Table 7.
Approaches to Treatment of Orthostatic Hypotension
| Treatment | Dose Regimen | Possible Side Effects |
|---|---|---|
|
| ||
| Support garments | ||
|
| ||
| Behavioral advice | ||
| Avoid sudden changes of body posture | ||
| Avoid hot baths and predisposing medications | ||
| Eat frequent small meals | ||
| Take insulin injections lying down | ||
|
| ||
| Fludrocortisone | 0.1 mg titrated to 0.5 to 2.0 mg/day | May predispose to congestive heart failure, hypertension |
|
| ||
| Sympathomimetics | ||
| Ephedrine | 15 – 45 mg, TID | Sympathetic symptoms |
| Midodrine | 2.5 – 10 mg, TID | Fewer centrally-mediated side effects |
| Clonidine | 0.1 – 0.5 mg at bedtime | Hypotension |
|
| ||
| Octreotide | 0.1 – 0.5 μg/kg/day | Injection site pain, diarrhea |
Pharmacologic approaches may also be employed. The mineralicorticoid, fludrocortisone, together with supplementary salt increases plasma volume. Unfortunately, it is generally ineffective until edema develops, which carries a risk of causing congestive heart failure and hypertension (Freeman, 2007). The mixed adrenergic agonist, ephedrine, the α-1 adrenergic agonist, midodrine, and the α-2 adrenergic agonist, clonidine have each been found to be effective in some patients, but it is important to begin with a low dose and titrate upward to minimize the various symptoms associated with their use (Freeman, 2007; Simmons, 2002). Finally, the somatostatin analog, octreotide may also help some patients who experience particularly refractory orthostatic hypotension after eating (Administration, 1997).
Many gastrointestinal symptoms may accompany DAN. Among the most common conditions is gastroparesis (Sawicki, 1996). Gastroparesis should always be suspected in patients with erratic glucose control. Table 8 addresses treatment of gastroparesis. As for all neuropathies, initial treatment should focus on improved blood glucose control. This is particularly important for gastrointestinal manifestations of DAN because good glucose control can improve gastric motor function. Eating frequent small meals is recommended, and patients should limit dietary fat to < 40 g/day and avoid excessive dietary fiber which can lead to formation of bezoars. Diabetic gastroparesis can be treated with a variety of prokinetic agents and recommended dose regimens are shown below. Unfortunately, it is often observed that tachyphylaxis develops after the first few doses. Periodic withdrawal may restore responsiveness and should be tried in apparently refractory cases. In severe cases, jejunostomy may allow the stomach to “rest” until it recovers function.
Table 8.
Approaches to Treatment of Diabetic Gastroparesis
| Treatment | Dose Regimen | Possible Side Effects |
|---|---|---|
|
| ||
| Behavioral advice | ||
| Improve glycemic control | ||
| Eat frequent small meals | ||
| Reduce dietary fat (<40 g/day) | ||
| Reduce dietary fiber | ||
|
| ||
| Metoclopramide | 10 mg, 30–60 min ac | Galactorrhea, extrapyramidal symptoms |
|
| ||
| Erythromycin | 250 mg, 30 min ac | Abdominal cramps, nausea, diarrhea, rash |
|
| ||
| Jujunostomy and liquid diet | ||
Diabetic diarrhea is also very common, usually characterized by an intermittent pattern. Table 9 provides approaches to evaluation and treatment of diarrhea in diabetic patients. The first step in treating diabetic diarrhea is excluding a treatable underlying cause. Drug-related diarrhea (due to metformin or acarbose treatment) and lactose intolerance should be considered. A hydrogen breath test can establish whether the diarrhea is due to bacterial overgrowth resulting from hypomotility. Diarrhea that resolves with fasting may be osmotic, caused by ingested substances, whereas diarrhea that continues when the patient is fasting suggests that a secretory process is involved, and neuroendocrine causes should be considered. Treatment, as with all manifestations of neuropathy should begin with good glycemic control. A broad-spectrum antibiotic such as metronidazole can be used to treat diarrhea resulting from bacterial overgrowth. Clonidine may improve diarrhea by reversing adrenergic overactivity. Cholestyramine may be used to chelate bile salts if the hydrogen breath test is normal and the patient fails to respond to antibiotics. Loperamide can be used to reduce the number of stools, but it should be used with caution due to risk of toxic megacolon. Diarrhea resistant to the aforementioned approaches may respond to octreotide.
Table 9.
Approaches to Treatment of Diabetic Diarrhea
| Treatment | Dose Regimen | Possible Side Effects |
|---|---|---|
|
| ||
| Exclude other underlying causes: | ||
| Bacterial overgrowth | ||
| Drug-related (acarbose, metformin, lactose intolerance) | ||
| Osmotic (resolves with fasting) | ||
| Secretory (consider neuroendocrine tumors) | ||
|
| ||
| Metronodiazole | 250 mg, TID, ≥ 3 wk | Fungal overgrowth |
|
| ||
| Clonidine | 0.1 mg, BID or TID | Orthostatic hypotension |
|
| ||
| Loperamide | 2 mg, QID | Toxic megacolon |
|
| ||
| Cholestyramine | 4 g, 1 to 6 times daily | |
|
| ||
| Octreotide | 50 μg, TID | Aggravated nutrient malabsorption |
DAN may also lead to bladder dysfunction. Diabetic cystopathy, or “neurogenic bladder” arises from loss of the afferent innervation of the bladder and accordingly, inability to sense a full bladder. As a result, voiding frequency is diminished and the patient may be unable to void completely. This predisposes a patient to recurrent urinary tract infections, overflow incontinence, and dribbling. Treatment of neurogenic bladder should begin with scheduled voiding, often coupled with manual pressure on the bladder to initiate urination (Credé maneuver). The parasympathomimetic agent, bethanechol (10 mg, QID) may be helpful, and extended sphincter relaxation can be achieved with the α-1 adrenergic antagonist, doxazosin (1–2 mg, BID or TID) (Freeman, 2007). Self-catheterization can be very useful and reduces the risk of urinary tract infections. Occasionally, chronic catheterization or transurethral surgery of the bladder neck may be necessary (Simmons, 2002).
Erectile dysfunction is one of the most common and distressing manifestations of DAN. The prevalence of erectile dysfunction in men with diabetes is exceedingly high; recently reported to be 67% in normotensive men with diabetes and 78% in men with both diabetes and hypertension (Giuliano, 2004). Erectile dysfunction may be the presenting symptom of diabetes, and importantly, it is a marker for the development of generalized vascular disease and for premature death from myocardial infarction (Freeman, 2007). Since erectile dysfunction may presage future cardiovascular events, a thorough cardiovascular evaluation should be performed on all diabetic men with erectile dysfunction. Erectile dysfunction can result from a variety of factors other than or in addition to DAN and it is useful to distinguish organic from psychogenic causes. Maintenance of nocturnal tumescence suggests a psychogenic cause, and erectile dysfunction due to DAN is nearly always accompanied by loss of ankle reflexes and reduced vibration sensation in the large toes. Treatment of erectile dysfunction should begin with optimization of glucose control and abstinence from alcohol and tobacco. A variety of prescription drugs can cause erectile dysfunction, and thus the physician should be aware of all medications the patient is receiving and attempt to eliminate those that can cause erectile dysfunction, or seek a substitute with less propensity to cause sexual dysfunction.
Phosphodiesterase inhibitors are now available, with different pharmacokinetic and side effect profiles to treat erectile dysfunction (Anderson, 2004). Sildenafil (50 mg, 60 minutes before sexual activity) or tadalafil (5 to 20 mg, 60 minutes before sexual activity) are effective in treating erectile dysfunction in men with diabetes (Safarinejad, 2004). However, all of these agents are contraindicated in patients being treated with nitroglycerine or other nitrate-containing drugs. For many men, injection of prostacyclin into the corpus cavernosum produces satisfactory erections, and surgically implanted penile prostheses are also available. Thus there are now several effective approaches to treating erectile dysfunction in men with diabetes.
5c. Causal Therapies
Topics covered previously in this review discussed proven therapies which either prevent or slow diabetic neuropathy (glycemic control) or diminish its effects (symptomatic therapies). As the only proven approach to treating the cause of diabetic neuropathy is glycemic control, pharmacological and neutraceutical therapies which aim to reverse diabetic neuropathy are examined here. These potential therapies attempt to mitigate the biochemical abberations which induce neuronal damage.
5C i. Polyol Pathway
The most common target of diabetic neuropathy therapies is sorbitol the first metabolic product of the polyol pathway, through the enzyme AR. AR converts cytosolic glucose to sorbitol by NADPH oxidation; SDH then converts sorbitol to fructose by NAD+ hydrogenation (Chung, 2005; Hers, 1956). Elevated sorbitol levels are thus indicators of hyperglycemia and good therapeutic targets for reducing oxidative stress resulting from excessive glucose metabolism.
Aldose Reductase Inhibitors
Aldose Reductase Inhibitors (ARIs) have historically been the primary target of diabetic neuropathy treatment, due largely to their success in reducing cataract-forming osmotic stress associated with polyol accumulation in the diabetic lens (Chylack, 1979; Kinoshita, 1968). Additionally, most studies of the human AR gene (AKR1B1) and its polymorphisms in diabetic patients indicate that the “high AR expression” genotype is correlated with elevated diabetic vascular complications and early diabetic neuropathy indicators, whereas the “low AR expression” genotype is correlated with diminished complications and indicators (Demaine, 2003; Donaghue, 2005; Thamotharampillai, 2006). Furthermore, ARIs have been successful in preventing and reversing nerve deterioration in rodent models (Cameron, 1986; Kato, 2000; Yagihashi, 1990). A variety of ARIs have entered the market; while most have effectively reduced nerve polyol levels, this result has not always translated to amelioration of diabetic neuropathy symptoms. Recent thinking posits that polyol levels themselves are not the best indicators of drug efficacy. Instead, the “Metabolic Flux Hypothesis” emphasizes cofactor turnover rate as a better marker of oxidative stress alleviation (Barnett, 1986; Cameron, 1994). This model suggests that much higher (~20–30 fold) doses of ARIs are needed to effectively decrease the rate of cofactor turnover and thereby alleviate common diabetic neuropathy symptoms (Oates, 2008, in press). Table 10 describes past and ongoing ARI trials and investigations.
Sorbinil
In 1981, Sorbinil was the prototype ARI to be developed solely for diabetic neuropathy treatment. Despite successfully reducing and preventing NCV deficits in rodent models, Sorbinil failed to produce noteworthy results in humans. While Sorbinil, a spiroimide, slightly improved in nerve fiber regeneration and human diabetic NCV (by 0.7–1.2 m/sec), it was associated with a high incidence of skin rash, and was withdrawn from the market in 1987 (Judzewitsch, 1983; Sima, 1988). However, Sorbinil’s moderate success paved the way for future ARI therapies.
Ponalrestat
Ponalrestat is a carboxylic acid that effectively lowers nerve sorbitol levels in vitro and in rats, but fails to do so in human diabetic nerves (Greene, 1993; Stribling, 1985). Ponalrestat is 99% plasma protein-bound in humans (~10-fold increase over rats), and most of the unbound acid is ionized at cellular pH. Such ions are slow to cross nerve plasma membranes, further diminishing Ponalrestat’s effectiveness.
Zopolrestat
Zopolrestat is a carboxylic acid analog of Ponalrestat that dose-dependently decreased diabetic rat nerve sorbitol and fructose levels. In human studies, low levels (250–500mg) of Zopolrestat decreased nerve sorbitol levels, but had no effect on fructose levels or symptom alleviation, and showed little NCV improvement. At higher levels (1000mg), Zopolrestat was significantly more effective at increasing NCV, but was associated with a higher incidence (7%) of elevated liver enzymes and eventually withdrawn (Arezzo, 1996). These trials illustrate that nerve sorbitol level per se is not the best indicator of nerve health, and that elevated ARI doses are likely needed to achieve significant diabetic neuropathy symptom improvement.
Zenarestat
Zenarestat is a carboxylic acid ARI also showing a dose-dependant increase of NCV; notably, higher doses of Zenarestat continue to improve NCV, increase nerve fiber density and nerve health even after nerve sorbitol levels have stabilized (Greene, 1999). Though its development was terminated due to a high incidence of elevated serum creatine levels, the studies demonstrated nonetheless that sorbitol is not the best marker of drug efficacy, and provides hope that at high doses ARIs can be more effective than previously thought (Gabbay, 2004).
AS-3201
AS-3201, or Ranirestat, is a well-tolerated spirosuccinimide discovered in 1998. Phase 2 trials were promising, showing few side effects and marked improvement in both NCV deficit and diabetic neuropathy symptoms (Bril, 2006)(Eisai Co., 2007) However, definitive Phase 3 study conclusions could not be drawn as of July 2007 due to the trial’s unusually high placebo effect. AS-3201 development is ongoing, and researchers hope that continued study and increased dosage of Ranirestat will prove effective in future diabetic neuropathy treatment (Oates, 2008, in press).
Epalrestat
In 1992, Epalrestat entered the Japanese market as a carboxylic acid ARI with minimum side-effects, but without conclusive evidence of efficacy backed by a randomized, double-blind placebo-controlled study. From 1997–2003 such a study was conducted, and at slightly elevated doses (150mg), Epalrestat delayed nerve deterioration and alleviated many common diabetic neuropathy symptoms such as limb numbness and cramping (Hotta, 2006). Though these results have not been replicated, Epalrestat is now the standard drug therapy for diabetic neuropathy in Japan.
Myo-inositol
Myo-inositol is a naturally occurring secondary messenger involved in proper nerve function. Myo-inositol depletion is associated with decreased Na+-K+-ATPase function and decreased NCV, and has been implicated in early-stage diabetic neuropathy pathology (Sima, 1997). Evidence suggests that dietary myo-inositol supplements might slow diabetic neuropathy progression, though further study is needed to assess efficacy.
5c ii. Hexosamine Pathway
As described above, activation of the hexosamine pathway generates UDPGlcN-Ac, which modulates transcription factors and ultimately induces neurovascular insult. While ARIs directly target toxic pathways, modulation of the hexosamine pathway can redirect glycolytic flow away from subsequent deleterious pathways. This mode of action offers an intriguing possibility for altering pathways in metabolic disorders.
Benfotiamine
Benfotiamine is a fat-soluble analogue of thiamine/vitamin B1 that activates transketolase, an enzyme converting fructose-6 phosphate into pentose-5 phosphates (Figure 10). The reduced fructose-6 phosphate input decreases flux through the hexosamine pathway (as well as flux through the advanced glycation end product (AGE) and the diacylglycerol (DAG) protein kinase C (PKC) pathways)(Hammes, 2003). The increased flux away from the hexosamine pathway and into the pentose-5 phosphate pathway may offer an additional benefit: increased redox capacity. One of the products of the pentose phosphate pathway is NADPH, a prime reactant in the formation of the antioxidant glutathione. Since NADPH is depleted in the polyol pathway, benfotiamine holds the speculative possibility of diminishing the effects of this pathway as well. Benfotiamine has successfully inhibited these pathways and prevented diabetic retinopathy in animal models (Hammes, 2003). In humans, Benfotiamine has been shown to improve pain associated with diabetic neuropathy and to improve NCV in conjunction with vitamins B6 and B12 (Haupt, 2005; Stracke, 1996; Winkler, 1999). Benfotiamine is currently availible as a dietary supplement in the United States.
Figure 10. Diagram of Benfotiamine Effect on Biochemical Pathways in Diabetic Complications.

Excess glucose activates flux through hexosamine pathway creating UDPGlcNAc from F-6-P. UDPGlcNAc modifies transcription factors which lead to inflammation. Addition of benfotiamine, a thiamine analog activates transketolase (TK) which diverts substrate away from the hexosamine pathway and into the pentose phosphate pathway. Adapted from (Hammes, 2003).
5c. iii PKC Pathway
Ruboxistaurin
Ruboxistaurin is a PKC-β competitive inhibitor that has effectively managed many complications of diabetes in clinical trials. It has been particularly successful in reducing the progression of diabetic retinopathy, endothelial vasodilation, and (to a lesser extent) nephropathy (Aiello, 2006; Beckman, 2002; Ishii, 1996; Tuttle, 2005). However, trials of Ruboxistaurin’s effect on diabetic neuropathy have not shown significant improvement (Vinik, 2005). Ruboxistaurin was being developed for US marketing by Eli Lilly and was pending FDA approval as a pharmaceutical agent for diabetic retinopathy. However, the company withdrew its marketing application in March, 2007, and Ruboxistaurin’s fate is currently unclear.
5c. iv AGE-RAGE Pathway
Clearly, glycemic control is the primary means for decreasing AGE formation. Given that this may be difficult to achieve, prevention of RAGE activation is an important alternative therapeutic goal for diabetic neuropathy (Figure 11). Two possible approaches are feasible: to prevent the formation of AGEs or to block RAGE. Numerous compounds have been investigated for anti-glycation activity but their use in humans is still debatable. The following section describes compounds that have been assessed for the ability to decrease activity of the RAGE axis in diabetic neuropathy.
Figure 11. Therapeutic targets on the AGE-RAGE pathway to DN.

The process of AGE damage and RAGE activation offer multiple approaches for preventing neuronal damage, from preventing AGE formation to blocking downstream signaling cascades.
Aspirin
As already mentioned, aspirin (acetylsalicylic acid-an NSAID) is a commonly used analgesic although long-term use in diabetic patients should be weighted against possible gastrointestinal side effects. In diabetic patients on high doses of aspirin, such as those with rheumatoid arthritis, the incidence of retinopathy is decreased compared with age-matched controls not taking aspirin, which suggested that aspirin may protect against glycation (Cotlier, 1981). Indeed, aspirin reduces glycation in vitro, and in animal experiments, potentially by acetylation of amino groups (Blakytny, 1992). Other analgesics such as paracetamol and ibuprofen also protect against glycation but cannot acetylate proteins (Blakytny & Harding, 1992). Alternatively, it is possible that aspirin does not directly alter glycation, but inhibits glycoxidation and AGE-cross-link formation; therefore its effects may be because of its antioxidant capacity(Fu, 1994). Besides the analgesic effects of aspirin, studies indicate a reduced risk of cardiovascular events for diabetic patients on low dose aspirin(Hennekens, 2004).
Aminoguanidine
Aminoguanidine (also called pimagedine) is a nucleophilic hydrazine compound and has received the most attention as a potential anti-glycation drug (Thornalley, 2003). Initially, it was thought that aminoguanidine prevented AGE formation by blocking carbonyl groups on Amadori products, although it is now known to react with carbonyl groups from reducing sugars or 3-DG (Edelstein, 1992; Lewis, 2006). In diabetic animals, aminoguanidine reduces nephropathy (Soulis, 1996), retinopathy (Chibber, 1994; Hammes, 1995), and neuropathy in some but not all studies (Birrell, 2000; Miyauchi, 1996). Preliminary studies in diabetic patients showed that aminoguanidine therapy for 28 days reduces hemoglobin-derived AGEs (Hb-AGE) but does not alter levels of Amadori products (Makita, 1992). Three other phase II and III trials of aminoguanidine have been completed with nephropathy endpoints but produced no benefit. The last was discontinued because of side effects in patients, which include flu-like symptoms, gastrointestinal disturbances, and anemia (Bolton, 2004). Despite the earlier promising results with aminoguanidine, it is unlikely to be used for therapeutic purposes. However, studies on anti-glycation compounds like aminoguanidine have provided evidence for the involvement of AGEs in the pathogenesis of diabetic complications.
Phenacylthiazolium bromide
Compounds capable of cleaving AGE cross-links have been described, opening up the exciting possibility of reversing diabetic complications. These compounds include N-phenacylthiazolium bromide (PTB), which can cleave AGE-cross-links by a mechanism which is still unclear. PTB has been used to cleave AGE cross-links between albumin and collagen in vitro and recent studies in diabetic rats have shown that PTB can prevent or reverse the accumulation of AGEs in blood vessels (Cooper, 2000). However, another study found that although PTB can reduce model AGE cross-links in vitro, it does not reduce AGE cross-links formed in vivo (Yang, 2003). Whether AGE-cross-link breakers are useful in vivo will also depend on their long-term toxicity. Due to the unstable nature of PTB, analogues such as alagebrium chloride, also known as ALT-711, have been developed. This compound provides renoprotection in diabetic mice (Coughlan, 2007; Peppa, 2006). Patient trials to date have found that ALT-711 is well tolerated and produces significant vascular benefit in the elderly through decreased blood pressure and increased vascular elasticity (Little, 2005; Zieman, 2007). Effects on microvascular complications of diabetes including neuropathy are not yet completed.
Blocking RAGE
There is considerable interest in compounds capable of blocking the interaction between AGEs and RAGE. RAGE can be blocked by usage of soluble RAGE (sRAGE), which is the extracellular ligand-binding domain of RAGE or by use of antibodies capable of reacting with RAGE. Studies by Schmidt and coworkers have performed multiple studies in diabetic mouse models using RAGE knockout mice and mice treated with sRAGE or anti-RAGE (Hudson, 2004). They demonstrate: topical sRAGE improves wound healing (Wear-Maggitti, 2004), sRAGE decreases atherosclerosis in ApoE knockout mice (Bucciarelli, 2002), RAGE blockade prevents the final stages of diabetogenesis in non-obese diabetic mice (Chen, 2004), and that RAGE blockage prevented sensory deficits (Bierhaus, 2004). Therefore, blockage of RAGE may be an important mechanism to prevent diabetic complications and the Schmidt group is actively working on translation of sRAGE to a clinical trial.
5c. v PARP Inhibitors
As PARP mediates both neuronal dysfunction and inflammation, inhibition of PARP holds the potential of improving two aberrant causeways in diabetic neuropathy, making it a promising target. PARP inhibitors such as 1,5-isoquinolinediol and 3-aminobenzamide have successfully improved these PARP-mediated dysfunctions in STZ-induced diabetic rats (Ilnytska, 2006; Li, 2005; Obrosova, 2005a). Additionally, Nicotinamide (vitamin B3) has been shown to act as both a PARP inhibitor and antioxidant in rodents, improving the complications of early diabetic peripheral neuropathy (Stevens, 2007). Nicotinamide is an attractive potential therapeutic due to its limited side effects and toxicity (Gale, 2004). A combination therapy for diabetic neuropathy including nicotinamide, the xanthine oxidase inhibitor allopurinol, and the antioxidant DL-α-lipoic acid is currently in trial.
5c. vi Antioxidants
Given the known mechanisms leading to diabetic neuropathy, a logical therapeutic approach is to prevent oxidative stress by increasing antioxidant defense. Antioxidant defense arises from (Hogan, 2003) antioxidant enzymes that catalyze the removal of ROS antioxidant molecules that prevent the oxidation of other molecules (Dworkin, 2007a), usually because they are readily oxidized molecules that chelate transition metal ions so they cannot catalyze the generation of ROS in a cell (Dworkin, 2005). A major portion of the body’s antioxidant defense comes from dietary intake of micronutrient molecules that facilitate one or more of these three mechanisms. This makes oral antioxidants an attractive strategy for both prevention and treatment of diabetic neuropathy. Many clinical trials of antioxidant defense therapies have been completed, mostly using a high dose of a single antioxidant compound. The results of these trials have been largely negative, despite the strong rationale for this approach. We will discuss several of the lead candidate compounds and summarize other known trials in Table 11.
Table 11.
Antioxidant therapy in diabetic neuropathy
| Compound | Trials/Notes | References |
|---|---|---|
| Coenzyme Q10 | Cofactor that improves metabolism but also a potent antioxidant. | (Bonnefont-Rousselot, 2004; Eriksson, 1999) |
| Nicotinamide | (AKA vitamin B3) is a weak PARP inhibitor, antioxidant, and calcium modulator. Effective in experimental diabetes and currently in a type 1 diabetes patient trial. | (Bonnefont-Rousselot, 2004; Eriksson, 1999; Stevens, 2007) |
| Eugenol | From clove oil; both anit-inflammatory and antioxidant. Improves vascular and neural deficits in STZ-treated rats. | (Nangle, 2006) |
| Taurine | Plasma taurine is depleted in diabetic rats and replacement decreases hyperalgesia and other neural and vascular deficits. | (Li, 2006; Li, 2005; Pop-Busui, 2001) |
| U83836E | A synthetic ROS scavenger, effective against oxidative stress, and neurovascular deficits in rats. | (Sayyed, 2006) |
| Oleuropein | From olive leaf, decreases blood glucose as well as oxidative stress in alloxan-treated rabbit. | (Al-Azzawie, 2006) |
| Minerals | Metal ions including vanadium, chromium, magnesium, zinc, selenium, copper contribute to antioxidant defense. They may become depleted in diabetic patients and should be included in the diet. | (Bonnefont-Rousselot, 2004) |
| Vitamin C | While vitamin C does not improve diabetes complications when given alone, it is used in combination with vitamin E or other antioxidants, since it facilitates effective antioxidant recycling. | (Dorchy, 1999; Jacobs, 2002; Will, 1999) |
| Quercetin | A flavonoid that attenuates thermal hyperalgesia and cold allodynia in STZ-induced diabetic rats | (Anjaneyulu, 2004) |
| Melatonin | Plasma levels decrease in diabetic patients with complications, supplementation reverses antioxidant status deficits and prevents complications. May both activate antioxidant response and scavenge ROS. | (Anwar & Meki, 2003; O’Brien, 1986; Tutuncu, 2005) |
| Apocynin | Decreases oxidative stress by inhibition of NAD(P)H oxidase | (Cotter, 2003a) |
| Rutin | A polyphenol that may activate the antioxidant response. Prevents oxidative stress in diabetic rats. | (Je, 2002) |
| Dimethylthiourea | A hydroxyl radical scavenger, prevents diabetes-induced mechanical and thermal nociceptive sensitivity and never blood flow deficits. | (Cameron, 2001) |
| Evening primrose oil | Initially used for therapeutic benefit of polyunsaturated fatty acids, also decreases oxidative stress. Efficacy further enhanced by conjugation with an antioxidant. | (Cameron, 1996; Ford, 2001) |
| Nitecapone | Catechol-O-methyltransferase inhibitor with potent antioxidant properties. Had some ability to prevent diabetic mechanical hyperalgesia, but essentially ineffective in preventing the development of diabetic neuropathy | (Pertovaara, 2001) |
| Troglitazone | Used as an insulin sensitizer in diabetic patients, but also operates as an antioxidant. May improve peripheral neuropathy in STZ-induced diabetic rats irrespective of blood glucose concentrations. | (Qiang, 1998) |
| N-acetylcysteine | Precursor of GSH that increases tissue GSH improves motor nerve conduction velocity and decreases oxidative stress in STZ-treated rats | (Sagara, 1996) |
Vitamin E
Vitamin E is a fat-soluble compound that exists in 8 isoforms with varying biological activity (Traber, 1995). Blood levels of vitamin E can decrease under prolonged oxidative stress and in individuals who cannot absorb dietary fat (Triantafillidis, 1998), are on a low fat diet, or are zinc deficient (Bunk, 1989). α-Tocopherol is the most active isoform and is the most common dietary supplement. This compound has been broadly tested for its ability to prevent chronic diseases involving oxidative stress including cancer and diabetes complications. While small studies have indicated that high intake of vitamin E may decrease incidence of certain cancers, large studies generally do not support the findings. One study found that regular intake of high does of vitamin E for more than 10 years decreased the risk of death from bladder cancer (Jacobs, 2002). These studies demonstrate the safety of long-term use of vitamin E. In addition to potent antioxidant action, vitamin E can promote immune function, DNA repair, and metabolism (Traber, 1996; Wozniak, 2004). Chronic vitamin E administration improves the ratio of cardiac sympathetic to parasympathetic tone in patients with type 2 diabetes when given 600 mg/day for 4 mo (Manzella, 2001).
α-Lipoic Acid
Alpha-lipoic acid, also termed thioctic acid, is an antioxidant that is available for treatment of DPN in some countries (Ziegler, 1999b). It has the potent ability to scavenge ROS, regenerate other antioxidants, and chelate metal ions (Packer, 1997). Some randomized controlled clinical trials have shown that intravenous infusions of α-lipoic acid (600 mg daily, 5 days/week for 3 weeks) significantly improved sensory symptoms of DPN or the Neuropathic Impairment Score (Ziegler, 1999a). In another small study of oral α-lipoic acid (800 mg, QD) a small (non-significant) trend toward improvement in measures of cardiac autonomic neuropathy was reported (Ziegler, 1997). In a more recent open-label trial of 10 days intravenous administration followed by 50 days of oral treatment, α-lipoic acid was found to improve several manifestations of autonomic neuropathy (Tankova, 2004). Other clinical trials have been completed with varying doses and either parenteral or oral administration of the drug. In 1995, a review of current evidence assessed trials that met specific requirements of randomization, double masking and placebo-controlled design (Foster, 2007). The study concluded that α-lipoic acid should be considered as a treatment for diabetic neuropathy since parenteral supplementation improves neuropathic symptoms over 3 weeks and oral treatment improves neuropathic deficits and potentially sensory symptoms. Another trial that was ongoing at this time was the Neurological Assessment of Thioctic Acid in Neuropathy (NATHAN) I. This was a 4-year oral treatment in 460 patients in a multicenter evaluation. The results have now appeared in abstract form and conclude that α-lipoic acid is tolerated long-term. It improves some neuropathic deficits and symptoms but does not improve nerve conduction in mild and moderate diabetic neuropathy. Despite many positive findings, α-lipoic acid remains broadly used in Europe and underutilized in the U.S.A., though commercially available as a nutritional supplement.
Botanicals
It has long been recognized that natural dietary products including red grapes, evening primrose, and cruciferous vegetables increase antioxidant status. In addition to many botanical extracts possessing antioxidant capacity, several compounds are known to increase the expression of antioxidant genes such as glutathione S-transferase and NAD(P)H:quinone oxidoreductase 1 (Halat, 2003). These enzymes belong to the class of phase 2 enzymes under the regulation of a specific promoter consensus region known as the antioxidant response element (ARE) (Itoh, 1999). Many compounds can activate the transcription factor, known as nuclear factor-E2-related factor-2 (Nrf-2), that binds this promoter. Oxidative stress can activate the promoter via phosphorylation of the Nrf-2 cytoplasmic chaperone protein Keap-1 (Tamasi, 2004). Botanical compounds may activate Nrf-2 either by inducing mild intracellular oxidative stress, by directly activating Nrf-2, or by causing recruitment of Nrf-2 co-activators, such as small Maf proteins (Dhakshinamoorthy, 2000; Kang, 2005; Venugopal, 1998). Several botanical compounds so effectively activate the antioxidant response they are used to treat diseases with an oxidative component, particularly cancer and Alzheimer’s disease (Perry, 2007; Thimmulappa, 2002).
Resveratrol
The most widely assessed botanical compound is resveratrol, extracted from red grapes. Studies in STZ-treated rats demonstrated attenuation of thermal hyperalgesia and cold allodynia as well as decreases in oxidative stress DNA damage, and nerve conduction deficits (Kumar, 2007; Sharma, 2006). Similarly, in type 1 diabetic mice resveratrol prevents neuropathic pain (Sharma, 2007). Resveratrol is likely to provide additional therapeutic benefits in T2DM patients because it also activates the SIRT1 genes that regulate glucose metabolism and insulin sensitivity (Chen, 2007; Sun, 2007; Zang, 2006) Resveratrol was assessed in a trial examining aging and cardiovascular disease and produced positive results (Labinskyy, 2006). Although trials in diabetes are indicated by these findings, there are none on record to date. Other botanicals that activate the antioxidant response in experimental diabetes and in patients include extract of Tinospora cordifolia (Prince, 1999), curcumin (Osawa, 2005), garlic oil (Anwar, 2003), evening primrose oil (Ford, 2001; Halat & Dennehy, 2003), and sulphoraphane (Perry, 2007; Thimmulappa, 2002). A controlled diabetes patient trial has not been performed for any of these compounds.
5c. vii Targeting Vascular Disease-ARBs and ACE Inhibitors
Several drugs are in common use in T2DM for blood pressure, cardiovascular disease, and nephropathy. The first line of attack for these diabetes-related conditions is inhibition of angiotensin-converting enzyme (ACE) or of angiotensin receptor. Evaluation of the outcomes of clinical trials suggests that preventing one complication is likely to have a positive impact upon other complications (Podar, 2002). Specifically, prevention of cardiovascular disease is likely to prevent macro-and microvascular complications. Large-scale studies of the effects of ACE inhibitors or angiotensin receptor blockers, known as ARBs, have not been done, although some small studies and prospective assessments have been performed. In experimental diabetes, enalapril, an ACE inhibitor, or L-158809, an angiotensin II receptor blocker, decreases neurovascular deficits including blood flow and motor nerve conduction velocity (Coppey, 2006). Another ACE inhibitor, Perindopril, prevents photoreceptor loss, an indicator of neuropathy (Bui, 2003). In a small clinical study, the ACE inhibitor trandolapril produced a significant improvement in peripheral neuropathy (Malik, 2000). Another study treated long-term diabetic patients with DAN but not high blood pressure or arterial disease with the ACE inhibitor Quinapril and/or the ARB Losartan (Didangelos, 2006). Either drug alone improved DAN and cardiac function; in combination the improvement may have been greater.
5c. viii Neurotrophic factors
Peripheral nervous system injury in diabetes may be the result of both hyperglycemia and loss of neurotrophic support normally provided by insulin. This hypothesis is supported by reports of abnormal expression levels of growth factors in diabetes. Thus, there is growing interest in exploring the potential utility of NGFs, insulin, IGFs, and others neurotrophic factors in the treatment of diabetic neuropathy.
Insulin receptors are found in the PNS on Schwann cells, pericytes, endothelial cells, and neurons, especially sensory neurons (Brussee, 2004; Sugimoto, 2002; Sugimoto, 2000). Insulin-deficient rat models of diabetes appear to have more severely progressive neuropathy compared to T2DM models, suggesting insulin deficiency itself contributes to the development of neuropathy (Kamiya, 2005; Pierson, 2003). In vitro, insulin activates survival-promoting PI-3K/Akt signaling and neurite outgrowth in sensory neurons (Fernyhough, 1993; Huang, 2005; Recio-Pinto, 1986). Local delivery of insulin to the spinal cords of STZ-treated rats improves nerve condition velocity measurements, and low-dose systemic delivery at a level that does not reduce hyperglycemia is able to decrease signs of mitochondrial distress in sensory neurons (Brussee, 2004; Singhal, 1997; Toth, 2006).
C-peptide, long thought to be merely an inert peptide fragment byproduct of insulin synthesis, is now believed to have biological activity of its own, although little is known mechanistically as there is currently no identified receptor (Wahren, 2004). C-peptide deficiency is concomitant with insulinopenia in T1DM. When C-peptide is replaced in diabetic rats, a number of measures of peripheral nerve function improve. Proposed mechanisms for these observations include potentiation of insulin signaling, vasodilation via nitric oxide release, and stimulation of the release of other neurotrophic factors (Cotter, 2003b; Kamiya, 2006; Pierson, 2003; Sima, 2004; Sima, 2001; Zhang, 2007).
Insulin-like growth factors (IGFs) I and II have profound effects on nervous system development and survival, mediated through activation of the IGF-I receptor (IGF-IR) (Fernandez, 2007; Leinninger, 2005). IGFs and the IGF-IR are expressed throughout the developing and adult nervous system. IGFs have been reported to be reduced in some animal models of diabetes, although this varies and may be dependent upon the model, type of diabetes, and tissue examined (Craner, 2002; Ekstrom, 1989; Kamiya, 2006; Schmidt, 2003; Wuarin, 1994; Zhuang, 1997). A number of preclinical studies in diabetic rats suggest systemic or intrathecal IGF therapy can improve neuropathy (Brussee, 2004; Ishii, 1995; Lupien, 2003; Schmidt, 1999; Schmidt, 2000; Toth, 2006; Zhuang, 1997). Clinical use of IGFs may be complicated by their widespread systemic effects (Russo, 2005). Additionally, the complex system of IGF binding proteins (IGFBPs) may impact efficacy. The IGFBPs regulate IGF bioavailability, and studies attempting to establish the status if the IGFBPs in diabetes have been inconsistent (Busiguina, 2000; Crosby, 1992; Han, 2006).
The system of neurotrophins is critical for the development and maintenance of the PNS and CNS (Huang, 2003; Kaplan, 2000) and includes nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and neurotrophins (NT) 3–6. NGF is not required for sensory neuronal survival in the adult PNS, but it does regulate axonal sprouting and the phenotype of sensory neurons (Chudler, 1997; Lindsay, 1988; Schwartz, 1982). Thus, preclinical studies of NGF in diabetic rats resulted in improvements in both signaling outcomes of the NGF system and PNS function, as well as positive effects on myelination (Apfel, 1994; Christianson, 2007; Christianson, 2003; Diemel, 1994; Elias, 1998; Fernyhough, 1995b; Sango, 1994; Unger, 1998). Clinical studies have not progressed past phase 3, however, and there is developing interest in small molecule activators of trkA as a potentially more viable alternative approach (Apfel, 2002).
BDNF is expressed by both peripheral neurons and muscle, and its receptor, trkB, is found on motor neurons and some sensory neurons (McMahon, 1994). Retrograde transport of endogenous, but not exogenous, BDNF to the neuron cell bodies is impaired in diabetic rats, suggesting there are problems with the local supply of BDNF at the peripheral nerve terminals (Mizisin, 1999). Exogenous BDNF is protective to large myelinated sensory fibers in STZ rats, but not smaller fibers, which is consistent with the distribution of trkB expression (Calcutt, 1998; Elias, 1998; Mizisin, 1997).
Preclinical studies of exogenous NT-3 therapy in diabetic rats have had mixed results. One study found improvement in large sensory fibers, but not motor fibers (Mizisin, 1998). Another study found effects on both large sensory and motor fibers (Mizisin, 2004). Intrathecal NT-3 increased myelinated fibers in the skin of diabetic mice, but without noticeable improvement of function (Christianson, 2007).
Ciliary derived neurotrophic factor (CNTF) is a cytokine with numerous neurotrophic properties (Vergara, 2004). It is only expressed in Schwann cells in the peripheral nervous system (Kobayashi, 2000), and levels of CNTF are reduced in diabetic rats (Calcutt, 1992). This deficiency can be improved by ARI therapy (Mizisin, 1997). Exogenous CNTF itself has therapeutic benefit in diabetic rats, improving function in both sensory and motor fibers, along with increasing regenerative capabilities (Calcutt, 2004; Mizisin, 2004). Therapeutic use of CNTF is complicated by its systemic effects, particularly on muscle, and thus more targeted delivery approaches are being considered (Bongioanni, 2004).
6. The Search for Novel Therapeutic Targets
While this review has discussed the fusion of over 30 years of research in diabetic neuropathy, glucose control alone still remains the only disease-modifying therapy for diabetic neuropathy (Leinninger, 2006b; Little, 2007; Vincent, 2008). This lack of progress, despite intense research, suggests that a new paradigm is needed. We contend that a “discovery approach” using informatics to analyze genomic and proteomic data from animal models and patients with DPN may provide much needed insight into disease pathogenesis and identify viable targets for disease modifying treatments. To date, only one animal study (and no human studies) has addressed alterations in peripheral nervous system gene expression in response to diabetes. Price and colleagues performed microarray analyses on Wistar rat dorsal root ganglion neurons, 1, 4, and 8 weeks post STZ-induced diabetes (Price, 2006). Nerve conduction measures confirmed the presence of DPN in these animals at weeks 4 and 8 (Price, 2006). At week 1, genes involved in glucose metabolism were upregulated, and by week 4 glutathione transferase was upregulated as would be predicted secondary to conditions of oxidative stress (Price, 2006).
What is now required is a comprehensive effort to establish the molecular signatures of neural tissues from genome wide screening of RNA from different animal models without and with DPN and DAN (Sullivan, 2007; Sullivan, 2008). A discovery approach for diabetic neuropathy is presented in Figure 12. Genome wide expression profiling of sensory and sympathetic neurons and nerves from different animal models of DPN and DAN will yield differentially regulated transcripts. These expression profiles will provide the data needed to define categories of genes that are functionally regulated in diabetic neuropathy; possible examples would be genes involved in metabolism or mitochondrial function or nerve regeneration. Further analyses of these data can define shared promoter modules among members of different gene categories, providing one or more specific targets for disease regulation (Ashburner, 2000). In parallel, an informatics approach can also define relevant pathways related to functional gene categories, providing additional information on novel mechanisms of disease pathogenesis or identifying new disease targets (Dennis, 2003) (Figure 12). The feasibility, power, and utility of using a discovery/informatics approach to uncover disease mechanisms is well described in chronic kidney disease in a series of papers by Kretzler and colleagues (Cohen, 2006; Schmid, 2003a; Schmid, 2003b; Schmid, 2006). In these studies, human renal biopsies were examined by Affymetrix™ microarray analysis and real time RT-PCR. The genes that were found to be differentially expressed between healthy and diseased tissue were put into the context of cellular pathways and used to predict regulatory elements controlling the observed changes. Regulatory elements identified could be used to identify drug targets and also to predict the downstream effects of gene expression, including the presence of biomarkers in chronic renal disease.
Figure 12.

Activities (in blue) are hypothesis driven and attempt to identify biomarkers based on the disequilibrium of identified targets in diabetic neuropathy, leading to an abnormal accumulation of products, such as modified proteins or small molecules. Activities in red are discovery oriented and seek to identify features of the data set that are predictive of diabetic neuropathy without necessarily corresponding to a single target.
As emphasized above, we contend a similar comprehensive approach is needed in diabetic neuropathy to further advance our understanding of disease pathogenesis and our development of disease modifying therapies. Of special interest is how this approach can in parallel lead to biomarker discovery. As outlined in Fig. 12, the first step is to employ microarrays and confirmatory Q-PCR to analyze gene expression of relevant neural tissue and use validated techniques to detect enriched pathways. These data in turn provide the knowledge to predict proteins and macromolecules influenced by gene expression. By using clustering and classification analysis, while maintaining high standards of mathematical validation throughout, one could discover those targets most likely to serve as useful biomarkers, such as modified proteins or small molecule products of targeted pathways. The data can also be analyzed to see if there is a constellation of genes that cluster in a unique way, suggesting a target not observed by the gene selection or pathway enrichment approach. A flow diagram of this type of analysis plan is presented in Figure 13 and is complimentary to the approach presented in Fig. 12.
Figure 13.

Discovery Approach for Novel Targets in Diabetic Neuropathy (DN). Genome wide expression profiling of neural tissues from animal models with diabetic neuropathy (DN) will yield differentially regulated transcripts. Analyses of these data using Gene Ontology (GO) will provide the data needed to define categories of genes that are functionally related providing a molecular signature for diabetic neuropathy. Further analyses of these data can define relevant pathways related to functional gene categories and shared promoter modules among members of different gene categories, providing one or more specific targets for disease regulation. These targets can be verified at the mRNA level, confirming the identification of a novel disease target.
7. Summary
Neuropathy is one of the most prevalent, devastating and costly complications of diabetes. Distal symmetrical sensorimotor polyneuropathy (DPN) is the leading cause of nontraumatic limb amputation. Diabetic autonomic neuropathy (DAN) can affect virtually any body system and is associated with greatly increased morbidity and mortality and can have a profound influence on quality of life. Several biochemical mechanisms of nerve and neurovascular damage have been identified and excessive production of reactive oxygen species, or “oxidative stress”, is thought to be a common etiologic factor. Diabetic neuropathies can be diagnosed with relatively simple tests of peripheral and autonomic nerve function. Treatment of diabetic neuropathy should always begin with efforts to optimize glycemic control and with patient education. There are now many useful pharmacologic approaches to treating painful neuropathy and most manifestations of autonomic neuropathy, but disease modifying treatments other than strict glycemic control await a more complete understanding of the underlying mechanisms of diabetic neuropathy and the development of pharmacologic agents based on this emerging knowledge.
Some positive results from preclinical studies with inhibitors implicated in the pathogenesis of diabetic neuropathy have been reported, but only time will tell whether and how these will translate into therapeutics. Discovery of these therapeutic targets will require implementation of bioinformatics, biochemistry, cellular biology, and physiology. It may be anticipated that with further progress toward validation of the recently proposed “unifying hypothesis”, development of agents to prevent mitochondrial oxidative damage will be the focus of intense study (Green, 2004).
Table 6.
Pharmaceutical therapies for diabetic neuropathy
| Drug | Number of patients | Daily Doses | Duration | Outcome | NNT | Reference |
|---|---|---|---|---|---|---|
| TCAs | ||||||
| Amitriptyline | 29 | ≤ 150mg | Cross over, 2X6 wks | Amitriptyline > placebo | 2.1 | (Max, 1987) |
| Desipramine | 20 | Average 201 mg | Cross over, 2X6 wks | Desipramine > placebo | 2.2 | (Max, 1992) |
| Imipramine | 19 | 25–350 mg | Cross over, 3X2 wks | Imipramine > placebo | 4 | (Sindrup, 1990b) |
| Clomipramine | 18 | 50–200 mg | Cross over, 3X2 wks | Clomipramine > placebo | 4.5 | (Sindrup, 1990c) |
| SSRIs | ||||||
| Paroxetine | 26 | 40 mg | Cross over, 3X2 wks | Paroxetine > placebo | 5 | (Sindrup, 1990a) |
| Citalopram | 18 | 40 mg | Cross over, 3X2 wks | Citalopram > placebo | 3 | (Sindrup, 1992) |
| Fluoxetine | 46 | 40 mg | Cross over, 2X6 wks | Fluoxetine = placebo | NA | (Max, 1992) |
| SNRIs | ||||||
| Venlafaxine | 244 | 150–225 mg | Parallel, 6 wks | Venlafaxine > placebo | 4.5 | (Rowbotham, 2004) |
| Duloxetine | 457 | 20, 60, 120 mg | Parallel, 12 wks | Duloxetine (60mg, 120 mg) > placebo | 60 mg: 4.3 120 mg: 3.8 |
(Goldstein, 2005) |
| Duloxetine | 348 | 60 mg, 120 mg | Parallel, 12 wks | Duloxetine (60mg, 120 mg > placebo | 60 mg: 11 120 mg: 5 |
(Raskin, 2005) |
| Duloxetine | 334 | 60 mg, 120 mg | Parallel, 12 wks | Duloxetine (60mg, 120 mg > placebo | 60 mg: 6.3 120 mg: 3.8 |
(Wernicke, 2006) |
| Anticonvulsants | ||||||
| Carbamazepine | 40 | 400 mg | Cross over, 2 wks | Carbamazepine > placebo | 2.3 | (Rull, 1969) |
| Lamotrigine | 59 | < 400 mg | Parallel, 8 wks | Lomotrigine > placebo | 4 | (Eisenberg, 2001) |
| Oxcarbazepine | 146 | < 1800 mg | Parallel, 16 wks | Oxcarbazepine > palcebo | 6 | (Dogra, 2005) |
| Topiramate | 323 | < 400 mg | Parallel, 12 wks | Topiramate > placebo | 7.4 | (Raskin, 2004) |
| Calcium channel α2- δagonists | ||||||
| Gabapentin | 165 | < 3600 mg | Parallel, 8 wks | Gabapentin > palcebo | 4 | (Backonja, 1998) |
| Pregabalin | 146 | 300 mg | Parallel, 8 wks | Pregabalin > placebo | 3.9 | (Rosenstock, 2004) |
| Pregabalin | 338 | 75, 300, 600 mg | Parallel, 5 wks | Pregabalin (300, 600 mg) > placebo | 300 mg: 3.6 600 mg: 3.3 |
(Lesser, 2004) |
| Pregabalin | 246 | 150, 600 mg | Parallel, 6 wks | Pregabalin (600 mg) > placebo | 600 mg: 4.2 | (Richter, 2005) |
| μ receptor agonists | ||||||
| Tramadol | 127 | 100–400 mg | Parallel, 6 wks | Tramadol > placebo | 3.1 | (Harati, 1998) |
| Oxycodon CR | 159 | 10–100 mg | Parallel, 6 wks | Oxycodon > placebo | NA | (Gimbel, 2003) |
| NMDA antagonists | ||||||
| Dextromethorphan | 19 | 400 mg | Cross over, 9 wks | Dextromethor phan > placebo | 3.2 | (Sang, 2002) |
| Memantine | 19 | 55 mg | Cross over, 9 wks | Memantine = placebo | NS | (Sang, 2002) |
| Topical agents | ||||||
| Capsaicin cream | 252 | 0.075% qid | 8 wks | Capsaicin > placebo | NA | (The Capsaicin Study, 1992) |
TCA: Tricyclic and tetracyclic antidepressants, SSRIs: Selective serotonin reuptake inhibitors, SNRIs: Serotonin-Norepinephrine Reuptake Inhibitors, NNT: number needed to treat, NA: not available. NS: not significant, Qid: four times a day.
Acknowledgments
We would like to thank Ms. Julie Erwin for her expert secretarial assistance. This was work was supported by NIH T32 D07245, the Juvenile Diabetes Research Foundation Center for the Study of Complications in Diabetes and the Program for Neurological Research and Development (PNRD).
Abbreviations
- ACE
angiotensin-converting enzyme
- AFT
Autonomic function tests
- AGE
Advanced glycation endproducts
- AR
Aldose reductase
- ARB
angiotensin receptor blocker
- ARI
Aldose reductase inhibitor
- BDNF
Brain-derived neurotrophic factor
- CAN
Cardiac autonomic neuropathy
- CGM
Continuous glucose monitor
- CNTF
Ciliary derived neurotrophic factor
- COX-2
Cyclooxygenase-2
- DAN
diabetic autonomic neuropathy
- DCCT
Diabetes control and complications trial
- DPN
diabetic polyneuropathy
- DRG
Dorsal root ganglion
- Drp1
Dynamin related protein 1
- EDIC
Epidemiology of diabetes interventions and complications
- EDX
Electrodiagnostic studies
- HSP
Heat shock protein
- IGF
Insulin like growth factor
- IGFBP
IGF binding protein
- MDNS
Michigan diabetic neuropathy score
- MNSI
Michigan neuropathy screening instrument
- Mt
Mitochondria
- NCV
Nerve conduction velocity
- NF-κB
Nuclear factor kappa B
- NGF
Nerve growth factor
- NMDA
N-methyl-d-aspartate
- NO
Nitric oxide
- NSAID
Non-steroidal anti-inflammatory drugs
- NT-3
Neurotrophin 3
- PAI-1
plasminogen activator inhibitor-1
- PARP
Poly(ADP-ribose) polymerase
- PKC
Protein kinase C
- QST
Quantitative sensory tests
- RAGE
Receptor for advanced glycation endproducts
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- SDH
Sorbitol dehydrogenase
- SNRI
Serotonin-Norepinephrine Reuptake Inhibitors
- sRAGE
soluble RAGE
- SSRI
Selective serotonin reuptake inhibitors
- STZ
Streptozotocin
- T1DM
Type 1 diabetes mellitus
- T2DM
Type 2 diabetes mellitus
- TCA
Tricylic and tetracylic antidepressants
- TGF-β1
Transforming growth factor β1
- VEGF
Vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abuaisha BB, Costanzi JB, Boulton AJ. Acupuncture for the treatment of chronic painful peripheral diabetic neuropathy: a long-term study. Diabetes research and clinical practice. 1998;39(2):115–121. doi: 10.1016/s0168-8227(97)00123-x. [DOI] [PubMed] [Google Scholar]
- Adler AI, Boyko EJ, Ahroni JH, Stensel V, Forsberg RC, Smith DG. Risk factors for diabetic peripheral sensory neuropathy. Results of the Seattle Prospective Diabetic Foot Study. Diabetes care. 1997;20(7):1162–1167. doi: 10.2337/diacare.20.7.1162. [DOI] [PubMed] [Google Scholar]
- Administration FaD. New drug application for Trovan (trovafloxacin) to the FDA. [Application Number: 020759/020760] Center for Drug Evaluation and Research. 1997:168–208. [Google Scholar]
- Adriaensen H, Plaghki L, Mathieu C, Joffroy A, Vissers K. Critical review of oral drug treatments for diabetic neuropathic pain-clinical outcomes based on efficacy and safety data from placebo-controlled and direct comparative studies. Diabetes/metabolism research and reviews. 2005;21(3):231–240. doi: 10.1002/dmrr.552. [DOI] [PubMed] [Google Scholar]
- Ahmed N. Advanced glycation endproducts--role in pathology of diabetic complications. Diabetes research and clinical practice. 2005;67(1):3–21. doi: 10.1016/j.diabres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Aiello LP, Clermont A, Arora V, Davis MD, Sheetz MJ, Bursell SE. Inhibition of PKC beta by oral administration of ruboxistaurin is well tolerated and ameliorates diabetes-induced retinal hemodynamic abnormalities in patients. Investigative ophthalmology & visual science. 2006;47(1):86–92. doi: 10.1167/iovs.05-0757. [DOI] [PubMed] [Google Scholar]
- Al-Azzawie HF, Alhamdani MS. Hypoglycemic and antioxidant effect of oleuropein in alloxan-diabetic rabbits. Life sciences. 2006;78(12):1371–1377. doi: 10.1016/j.lfs.2005.07.029. [DOI] [PubMed] [Google Scholar]
- Anderson PC, Gommersall L, Hayne D, Arya M, Patel HR. New phosphodiesterase inhibitors in the treatment of erectile dysfunction. Expert opinion on pharmacotherapy. 2004;5(11):2241–2249. doi: 10.1517/14656566.5.11.2241. [DOI] [PubMed] [Google Scholar]
- Anjaneyulu M, Chopra K. Quercetin attenuates thermal hyperalgesia and cold allodynia in STZ-induced diabetic rats. Indian journal of experimental biology. 2004;42(8):766–769. [PubMed] [Google Scholar]
- Anwar MM, Meki AR. Oxidative stress in streptozotocin-induced diabetic rats: effects of garlic oil and melatonin. Comparative biochemistry and physiology. 2003;135(4):539–547. doi: 10.1016/s1095-6433(03)00114-4. [DOI] [PubMed] [Google Scholar]
- Apfel SC. Nerve growth factor for the treatment of diabetic neuropathy: what went wrong, what went right, and what does the future hold? International review of neurobiology. 2002;50:393–413. doi: 10.1016/s0074-7742(02)50083-0. [DOI] [PubMed] [Google Scholar]
- Apfel SC, Arezzo JC, Brownlee M, Federoff H, Kessler JA. Nerve growth factor administration protects against experimental diabetic sensory neuropathy. Brain research. 1994;634(1):7–12. doi: 10.1016/0006-8993(94)90252-6. [DOI] [PubMed] [Google Scholar]
- Arezzo J, Klioze S, Peterson M, Lakshminarayanan M. Zopolrestat Phase II Neuropathy Study Group: Efficacy and Safety Results of a Phase II Multicenter Study of the Aldose Reductase Inhibitor Zopolrestat in Patients with Peripheral Symmetrical Diabetic Polyneuropathy. Diabetes care. 1996;45(2):276A. [Google Scholar]
- Arikawa E, Ma RC, Isshiki K, Luptak I, He Z, Yasuda Y, Maeno Y, Patti ME, Weir GC, Harris RA, Zammit VA, Tian R, King GL. Effects of insulin replacements, inhibitors of angiotensin, and PKCbeta’s actions to normalize cardiac gene expression and fuel metabolism in diabetic rats. Diabetes. 2007;56(5):1410–1420. doi: 10.2337/db06-0655. [DOI] [PubMed] [Google Scholar]
- Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP, Estaquier J, Sheng M, Blackstone C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol. 2005;15(23):2112–2118. doi: 10.1016/j.cub.2005.10.041. [DOI] [PubMed] [Google Scholar]
- Arrieta O, Garcia-Navarrete R, Zuniga S, Ordonez G, Ortiz A, Palencia G, Morales-Espinosa D, Hernandez-Pedro N, Sotelo J. Retinoic acid increases tissue and plasma contents of nerve growth factor and prevents neuropathy in diabetic mice. European journal of clinical investigation. 2005;35(3):201–207. doi: 10.1111/j.1365-2362.2005.01467.x. [DOI] [PubMed] [Google Scholar]
- Asbury AK, Porte D., Jr Report and recommnedations of the San Antoio conference on diabetic neuropathy. Diabetes. 1988;37:1000–1004. doi: 10.2337/diab.37.7.1000. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Association, A. D. Clinical practice recommendations 2004. American Diabetes Association; 2004. [Google Scholar]
- Atli A, Dogra S. Zonisamide in the treatment of painful diabetic neuropathy: a randomized, double-blind, placebo-controlled pilot study. Pain medicine (Malden, Mass. 2005;6(3):225–234. doi: 10.1111/j.1526-4637.2005.05035.x. [DOI] [PubMed] [Google Scholar]
- Averill S, McMahon SB, Clary DO, Reichardt LF, Priestley JV. Immunocytochemical localization of trkA receptors in chemically identified subgroups of adult rat sensory neurons. The European journal of neuroscience. 1995;7(7):1484–1494. doi: 10.1111/j.1460-9568.1995.tb01143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad N, Emanuele NV, Abraira C, Henderson WG, Colwell J, Levin SR, Nuttall FQ, Comstock JP, Sawin CT, Silbert C, Rubino FA. The effects of intensive glycemic control on neuropathy in the VA cooperative study on type II diabetes mellitus (VA CSDM) Journal of diabetes and its complications. 1999;13(5–6):307–313. doi: 10.1016/s1056-8727(99)00062-8. [DOI] [PubMed] [Google Scholar]
- Backonja M, Beydoun A, Edwards KR, Schwartz SL, Fonseca V, Hes M, LaMoreaux L, Garofalo E. Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus: a randomized controlled trial [see comments] Journal of the American Medical Association. 1998;280:1831–1836. doi: 10.1001/jama.280.21.1831. [DOI] [PubMed] [Google Scholar]
- Barbacid M. The Trk family of neurotrophin receptors. Journal of neurobiology. 1994;25(11):1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- Barbano RL, Herrmann DN, Hart-Gouleau S, Pennella-Vaughan J, Lodewick PA, Dworkin RH. Effectiveness, tolerability, and impact on quality of life of the 5% lidocaine patch in diabetic polyneuropathy. Archives of neurology. 2004;61(6):914–918. doi: 10.1001/archneur.61.6.914. [DOI] [PubMed] [Google Scholar]
- Barnett PA, Gonzalez RG, Chylack LT, Jr, Cheng HM. The effect of oxidation on sorbitol pathway kinetics. Diabetes. 1986;35(4):426–432. doi: 10.2337/diab.35.4.426. [DOI] [PubMed] [Google Scholar]
- Barrett AM, Lucero MA, Le T, Robinson RL, Dworkin RH, Chappell AS. Epidemiology, public health burden, and treatment of diabetic peripheral neuropathic pain: a review. Pain medicine (Malden, Mass. 2007;8(Suppl 2):S50–62. doi: 10.1111/j.1526-4637.2006.00179.x. [DOI] [PubMed] [Google Scholar]
- Bax G, Fagherazzi C, Piarulli F, Nicolucci A, Fedele D. Reproducibility of Michigan Neuropathy Screening Instrument (MNSI). A comparison with tests using the vibratory and thermal perception thresholds. Diabetes care. 1996;19(8):904–905. doi: 10.2337/diacare.19.8.904. [DOI] [PubMed] [Google Scholar]
- Beckman JA, Goldfine AB, Gordon MB, Garrett LA, Creager MA. Inhibition of protein kinase Cbeta prevents impaired endothelium-dependent vasodilation caused by hyperglycemia in humans. Circulation research. 2002;90(1):107–111. doi: 10.1161/hh0102.102359. [DOI] [PubMed] [Google Scholar]
- Beydoun A, Shaibani A, Hopwood M, Wan Y. Oxcarbazepine in painful diabetic neuropathy: results of a dose-ranging study. Acta neurologica Scandinavica. 2006;113(6):395–404. doi: 10.1111/j.1600-0404.2006.00631.x. [DOI] [PubMed] [Google Scholar]
- Bierhaus A, Haslbeck KM, Humpert PM, Liliensiek B, Dehmer T, Morcos M, Sayed AA, Andrassy M, Schiekofer S, Schneider JG, Schulz JB, Heuss D, Neundorfer B, Dierl S, Huber J, Tritschler H, Schmidt AM, Schwaninger M, Haering HU, Schleicher E, Kasper M, Stern DM, Arnold B, Nawroth PP. Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. The Journal of clinical investigation. 2004;114(12):1741–1751. doi: 10.1172/JCI18058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell AM, Heffernan SJ, Ansselin AD, McLennan S, Church DK, Gillin AG, Yue DK. Functional and structural abnormalities in the nerves of type I diabetic baboons: aminoguanidine treatment does not improve nerve function. Diabetologia. 2000;43(1):110–116. doi: 10.1007/s001250050014. [DOI] [PubMed] [Google Scholar]
- Blakytny R, Harding JJ. Prevention of cataract in diabetic rats by aspirin, paracetamol (acetaminophen) and ibuprofen. Experimental eye research. 1992;54(4):509–518. doi: 10.1016/0014-4835(92)90129-g. [DOI] [PubMed] [Google Scholar]
- Bolton WK, Cattran DC, Williams ME, Adler SG, Appel GB, Cartwright K, Foiles PG, Freedman BI, Raskin P, Ratner RE, Spinowitz BS, Whittier FC, Wuerth JP. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. American journal of nephrology. 2004;24(1):32–40. doi: 10.1159/000075627. [DOI] [PubMed] [Google Scholar]
- Bongioanni P, Reali C, Sogos V. Ciliary neurotrophic factor (CNTF) for amyotrophic lateral sclerosis/motor neuron disease. Cochrane database of systematic reviews (Online) 2004;(3):CD004302. doi: 10.1002/14651858.CD004302.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefont-Rousselot D. The role of antioxidant micronutrients in the prevention of diabetic complications. Treatments in endocrinology. 2004;3(1):41–52. doi: 10.2165/00024677-200403010-00005. [DOI] [PubMed] [Google Scholar]
- Boulton AJ. The diabetic foot: from art to science. The 18th Camillo Golgi lecture. Diabetologia. 2004a;47(8):1343–1353. doi: 10.1007/s00125-004-1463-y. [DOI] [PubMed] [Google Scholar]
- Boulton AJ, Kirsner RS, Vileikyte L. Clinical practice. Neuropathic diabetic foot ulcers. The New England journal of medicine. 2004b;351(1):48–55. doi: 10.1056/NEJMcp032966. [DOI] [PubMed] [Google Scholar]
- Boulton AJ, Levin S, Comstock J. A multicentre trial of the aldose-reductase inhibitor, tolrestat, in patients with symptomatic diabetic neuropathy. Diabetologia. 1990;33(7):431–437. doi: 10.1007/BF00404095. [DOI] [PubMed] [Google Scholar]
- Boulton AJ, Malik RA, Arezzo JC, Sosenko JM. Diabetic somatic neuropathies. Diabetes care. 2004c;27(6):1458–1486. doi: 10.2337/diacare.27.6.1458. [DOI] [PubMed] [Google Scholar]
- Boulton AJ, Vinik AI, Arezzo JC, Bril V, Feldman EL, Freeman R, Malik RA, Maser RE, Sosenko JM, Ziegler D. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes care. 2005;28(4):956–962. doi: 10.2337/diacare.28.4.956. [DOI] [PubMed] [Google Scholar]
- Bril V, Buchanan RA. Long-term effects of ranirestat (AS-3201) on peripheral nerve function in patients with diabetic sensorimotor polyneuropathy. Diabetes care. 2006;29(1):68–72. doi: 10.2337/diacare.29.01.06.dc05-1447. [DOI] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- Brownlee M, Hirsch IB. Glycemic variability: a hemoglobin A1c-independent risk factor for diabetic complications. Jama. 2006;295(14):1707–1708. doi: 10.1001/jama.295.14.1707. [DOI] [PubMed] [Google Scholar]
- Brussee V, Cunningham FA, Zochodne DW. Direct insulin signaling of neurons reverses diabetic neuropathy. Diabetes. 2004;53(7):1824–1830. doi: 10.2337/diabetes.53.7.1824. [DOI] [PubMed] [Google Scholar]
- Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, Goova MT, Moser B, Kislinger T, Lee DC, Kashyap Y, Stern DM, Schmidt AM. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 2002;106(22):2827–2835. doi: 10.1161/01.cir.0000039325.03698.36. [DOI] [PubMed] [Google Scholar]
- Bui BV, Armitage JA, Tolcos M, Cooper ME, Vingrys AJ. ACE inhibition salvages the visual loss caused by diabetes. Diabetologia. 2003;46(3):401–408. doi: 10.1007/s00125-003-1042-7. [DOI] [PubMed] [Google Scholar]
- Bunk MJ, Dnistrian AM, Schwartz MK, Rivlin RS. Dietary zinc deficiency decreases plasma concentrations of vitamin E. Proceedings of the Society for Experimental Biology and Medicine. Society for Experimental Biology and Medicine (New York, N Y. 1989;190(4):379–384. doi: 10.3181/00379727-190-42876. [DOI] [PubMed] [Google Scholar]
- Busiguina S, Fernandez AM, Barrios V, Clark R, Tolbert DL, Berciano J, Torres-Aleman I. Neurodegeneration is associated to changes in serum insulin-like growth factors. Neurobiology of disease. 2000;7(6 Pt B):657–665. doi: 10.1006/nbdi.2000.0311. [DOI] [PubMed] [Google Scholar]
- Cai F, Tomlinson DR, Fernyhough P. Elevated expression of neurotrophin–3 mRNA in sensory nerve of streptozotocin-diabetic rats. Neuroscience letters. 1999;263(2–3):81–84. doi: 10.1016/s0304-3940(99)00124-x. [DOI] [PubMed] [Google Scholar]
- Calcutt NA, Chen P, Hua XY. Effects of diabetes on tissue content and evoked release of calcitonin gene-related peptide-like immunoreactivity from rat sensory nerves. Neuroscience letters. 1998;254(3):129–132. doi: 10.1016/s0304-3940(98)00692-2. [DOI] [PubMed] [Google Scholar]
- Calcutt NA, Freshwater JD, Mizisin AP. Prevention of sensory disorders in diabetic Sprague-Dawley rats by aldose reductase inhibition or treatment with ciliary neurotrophic factor. Diabetologia. 2004;47(4):718–724. doi: 10.1007/s00125-004-1354-2. [DOI] [PubMed] [Google Scholar]
- Calcutt NA, Jolivalt CG, Fernyhough P. Growth factors as therapeutics for diabetic neuropathy. Current drug targets. 2008;9(1):47–59. doi: 10.2174/138945008783431727. [DOI] [PubMed] [Google Scholar]
- Calcutt NA, Muir D, Powell HC, Mizisin AP. Reduced ciliary neuronotrophic factor-like activity in nerves from diabetic or galactose-fed rats. Brain research. 1992;575(2):320–324. doi: 10.1016/0006-8993(92)90097-s. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Cotter MA. The relationship of vascular changes to metabolic factors in diabetes mellitus and their role in the development of peripheral nerve complications. Diabetes/metabolism reviews. 1994;10(3):189–224. doi: 10.1002/dmr.5610100302. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Cotter MA. Interaction between oxidative stress and gamma-linolenic acid in impaired neurovascular function of diabetic rats. The American journal of physiology. 1996;271(3 Pt 1):E471–476. doi: 10.1152/ajpendo.1996.271.3.E471. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Leonard MB, Ross IS, Whiting PH. The effects of sorbinil on peripheral nerve conduction velocity, polyol concentrations and morphology in the streptozotocin-diabetic rat. Diabetologia. 1986;29(3):168–174. doi: 10.1007/BF02427088. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Tuck Z, McCabe L, Cotter MA. Effect of the hydroxyl radical scavenger, dimethylthiourea, on peripheral nerve tissue perfusion, conduction velocity and nociception in experimental diabetes. Diabetologia. 2001;44(9):1161–1169. doi: 10.1007/s001250100626. [DOI] [PubMed] [Google Scholar]
- Chadda VS, Mathur MS. Double blind study of the effects of diphenylhydantoin sodium on diabetic neuropathy. The Journal of the Association of Physicians of India. 1978;26(5):403–406. [PubMed] [Google Scholar]
- Chen WP, Chi TC, Chuang LM, Su MJ. Resveratrol enhances insulin secretion by blocking K(ATP) and K(V) channels of beta cells. European journal of pharmacology. 2007;568(1–3):269–277. doi: 10.1016/j.ejphar.2007.04.062. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yan SS, Colgan J, Zhang HP, Luban J, Schmidt AM, Stern D, Herold KC. Blockade of late stages of autoimmune diabetes by inhibition of the receptor for advanced glycation end products. J Immunol. 2004;173(2):1399–1405. doi: 10.4049/jimmunol.173.2.1399. [DOI] [PubMed] [Google Scholar]
- Cheng C, Zochodne DW. Sensory neurons with activated caspase-3 survive long-term experimental diabetes. Diabetes. 2003;52(9):2363–2371. doi: 10.2337/diabetes.52.9.2363. [DOI] [PubMed] [Google Scholar]
- Chibber R, Molinatti PA, Wong JS, Mirlees D, Kohner EM. The effect of aminoguanidine and tolrestat on glucose toxicity in bovine retinal capillary pericytes. Diabetes. 1994;43(6):758–763. doi: 10.2337/diab.43.6.758. [DOI] [PubMed] [Google Scholar]
- Christianson JA, Ryals JM, Johnson MS, Dobrowsky RT, Wright DE. Neurotrophic modulation of myelinated cutaneous innervation and mechanical sensory loss in diabetic mice. Neuroscience. 2007;145(1):303–313. doi: 10.1016/j.neuroscience.2006.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JA, Ryals JM, McCarson KE, Wright DE. Beneficial actions of neurotrophin treatment on diabetes-induced hypoalgesia in mice. J Pain. 2003;4(9):493–504. doi: 10.1016/j.jpain.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Chudler EH, Anderson LC, Byers MR. Nerve growth factor depletion by autoimmunization produces thermal hypoalgesia in adult rats. Brain research. 1997;765(2):327–330. doi: 10.1016/s0006-8993(97)00681-1. [DOI] [PubMed] [Google Scholar]
- Chung SS, Chung SK. Aldose reductase in diabetic microvascular complications. Current drug targets. 2005;6(4):475–486. doi: 10.2174/1389450054021891. [DOI] [PubMed] [Google Scholar]
- Chylack LT, Jr, Henriques HF, 3rd, Cheng HM, Tung WH. Efficacy of Alrestatin, an aldose reductase inhibitor, in human diabetic and nondiabetic lenses. Ophthalmology. 1979;86(9):1579–1585. doi: 10.1016/s0161-6420(79)35364-7. [DOI] [PubMed] [Google Scholar]
- Clarke P, Gray A, Holman R. Estimating utility values for health states of type 2 diabetic patients using the EQ-5D (UKPDS 62) Med Decis Making. 2002;22(4):340–349. doi: 10.1177/0272989X0202200412. [DOI] [PubMed] [Google Scholar]
- Cohen CD, Klingenhoff A, Boucherot A, Nitsche A, Henger A, Brunner B, Schmid H, Merkle M, Saleem MA, Koller KP, Werner T, Grone HJ, Nelson PJ, Kretzler M. Comparative promoter analysis allows de novo identification of specialized cell junction-associated proteins. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(15):5682–5687. doi: 10.1073/pnas.0511257103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JA, Jeffers BW, Faldut D, Marcoux M, Schrier RW. Risks for sensorimotor peripheral neuropathy and autonomic neuropathy in non-insulin-dependent diabetes mellitus (NIDDM) Muscle & nerve. 1998;21(1):72–80. doi: 10.1002/(sici)1097-4598(199801)21:1<72::aid-mus10>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Cohen KL, Harris S. Efficacy and safety of nonsteroidal anti-inflammatory drugs in the therapy of diabetic neuropathy. Archives of internal medicine. 1987;147:1442–1444. [PubMed] [Google Scholar]
- Collins SL, Moore RA, McQuayHj &, Wiffen P. Antidepressants and anticonvulsants for diabetic neuropathy and postherpetic neuralgia: a quantitative systematic review. Journal of pain and symptom management. 2000;20(6):449–458. doi: 10.1016/s0885-3924(00)00218-9. [DOI] [PubMed] [Google Scholar]
- Conti G, Scarpini E, Baron P, Livraghi S, Tiriticco M, Bianchi R, Vedeler C, Scarlato G. Macrophage infiltration and death in the nerve during the early phases of experimental diabetic neuropathy: a process concomitant with endoneurial induction of IL-1beta and p75NTR. Journal of the neurological sciences. 2002;195(1):35–40. doi: 10.1016/s0022-510x(01)00684-0. [DOI] [PubMed] [Google Scholar]
- Cooper ME, Thallas V, Forbes J, Scalbert E, Sastra S, Darby I, Soulis T. The cross-link breaker, N-phenacylthiazolium bromide prevents vascular advanced glycation end-product accumulation. Diabetologia. 2000;43(5):660–664. doi: 10.1007/s001250051355. [DOI] [PubMed] [Google Scholar]
- Coppey LJ, Davidson EP, Rinehart TW, Gellett JS, Oltman CL, Lund DD, Yorek MA. ACE inhibitor or angiotensin II receptor antagonist attenuates diabetic neuropathy in streptozotocin-induced diabetic rats. Diabetes. 2006;55(2):341–348. doi: 10.2337/diabetes.55.02.06.db05-0885. [DOI] [PubMed] [Google Scholar]
- Coppini DV, Bowtell PA, Weng C, Young PJ, Sonksen PH. Showing neuropathy is related to increased mortality in diabetic patients - a survival analysis using an accelerated failure time model. Journal of clinical epidemiology. 2000;53(5):519–523. doi: 10.1016/s0895-4356(99)00170-5. [DOI] [PubMed] [Google Scholar]
- Cortright RN, Azevedo JL, Jr, Zhou Q, Sinha M, Pories WJ, Itani SI, Dohm GL. Protein kinase C modulates insulin action in human skeletal muscle. American journal of physiology. 2000;278(3):E553–562. doi: 10.1152/ajpendo.2000.278.3.E553. [DOI] [PubMed] [Google Scholar]
- Cotlier E. Aspirin effect on cataract formation in patients with rheumatoid arthritis alone or combined with diabetes. International ophthalmology. 1981;3(3):173–177. doi: 10.1007/BF00130701. [DOI] [PubMed] [Google Scholar]
- Cotter MA, Cameron NE. Effect of the NAD(P)H oxidase inhibitor, apocynin, on peripheral nerve perfusion and function in diabetic rats. Life sciences. 2003a;73(14):1813–1824. doi: 10.1016/s0024-3205(03)00508-3. [DOI] [PubMed] [Google Scholar]
- Cotter MA, Ekberg K, Wahren J, Cameron NE. Effects of proinsulin C-peptide in experimental diabetic neuropathy: vascular actions and modulation by nitric oxide synthase inhibition. Diabetes. 2003b;52(7):1812–1817. doi: 10.2337/diabetes.52.7.1812. [DOI] [PubMed] [Google Scholar]
- Coughlan MT, Forbes JM, Cooper ME. Role of the AGE crosslink breaker, alagebrium, as a renoprotective agent in diabetes. Kidney international. 2007;(106):S54–60. doi: 10.1038/sj.ki.5002387. [DOI] [PubMed] [Google Scholar]
- Craner MJ, Klein JP, Black JA, Waxman SG. Preferential expression of IGF-I in small DRG neurons and down-regulation following injury. Neuroreport. 2002;13(13):1649–1652. doi: 10.1097/00001756-200209160-00016. [DOI] [PubMed] [Google Scholar]
- Crosby SR, Tsigos C, Anderton CD, Gordon C, Young RJ, White A. Elevated plasma insulin-like growth factor binding protein-1 levels in type 1 (insulin-dependent) diabetic patients with peripheral neuropathy. Diabetologia. 1992;35(9):868–872. doi: 10.1007/BF00399934. [DOI] [PubMed] [Google Scholar]
- Currie CJ, Poole CD, Woehl A, Morgan CL, Cawley S, Rousculp MD, Covington MT, Peters JR. The financial costs of healthcare treatment for people with Type 1 or Type 2 diabetes in the UK with particular reference to differing severity of peripheral neuropathy. Diabet Med. 2007;24(2):187–194. doi: 10.1111/j.1464-5491.2006.02057.x. [DOI] [PubMed] [Google Scholar]
- Dagogo-Jack SE, Craft S, Cryer PE. Hypoglycemia-associated autonomic failure in insulin-dependent diabetes mellitus. Recent antecedent hypoglycemia reduces autonomic responses to, symptoms of, and defense against subsequent hypoglycemia. The Journal of clinical investigation. 1993;91(3):819–828. doi: 10.1172/JCI116302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallocchio C, Buffa C, Mazzarello P, Chiroli S. Gabapentin vs. amitriptyline in painful diabetic neuropathy: an open-label pilot study. Journal of pain and symptom management. 2000;20(4):280–285. doi: 10.1016/s0885-3924(00)00181-0. [DOI] [PubMed] [Google Scholar]
- Das Evcimen N, King GL. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res. 2007;55(6):498–510. doi: 10.1016/j.phrs.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Delcroix JD, Michael GJ, Priestley JV, Tomlinson DR, Fernyhough P. Effect of nerve growth factor treatment on p75NTR gene expression in lumbar dorsal root ganglia of streptozocin-induced diabetic rats. Diabetes. 1998;47(11):1779–1785. doi: 10.2337/diabetes.47.11.1779. [DOI] [PubMed] [Google Scholar]
- Delcroix JD, Tomlinson DR, Fernyhough P. Diabetes and axotomy-induced deficits in retrograde axonal transport of nerve growth factor correlate with decreased levels of p75LNTR protein in lumbar dorsal root ganglia. Brain Res Mol Brain Res. 1997;51(1–2):82–90. doi: 10.1016/s0169-328x(97)00215-5. [DOI] [PubMed] [Google Scholar]
- Demaine AG. Polymorphisms of the aldose reductase gene and susceptibility to diabetic microvascular complications. Current medicinal chemistry. 2003;10(15):1389–1398. doi: 10.2174/0929867033457359. [DOI] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome biology. 2003;4(5):P3. [PubMed] [Google Scholar]
- Dhakshinamoorthy S, Long DJ, 2nd, Jaiswal AK. Antioxidant regulation of genes encoding enzymes that detoxify xenobiotics and carcinogens. Current topics in cellular regulation. 2000;36:201–216. doi: 10.1016/s0070-2137(01)80009-1. [DOI] [PubMed] [Google Scholar]
- Didangelos TP, Arsos GA, Karamitsos DT, Athyros VG, Georga SD, Karatzas ND. Effect of quinapril or losartan alone and in combination on left ventricular systolic and diastolic functions in asymptomatic patients with diabetic autonomic neuropathy. Journal of diabetes and its complications. 2006;20(1):1–7. doi: 10.1016/j.jdiacomp.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Diemel LT, Brewster WJ, Fernyhough P, Tomlinson DR. Expression of neuropeptides in experimental diabetes; effects of treatment with nerve growth factor or brain-derived neurotrophic factor. Brain Res Mol Brain Res. 1994;21(1–2):171–175. doi: 10.1016/0169-328x(94)90391-3. [DOI] [PubMed] [Google Scholar]
- Dogra S, Beydoun S, Mazzola J, Hopwood M, Wan Y. Oxcarbazepine in painful diabetic neuropathy: a randomized, placebo-controlled study. European journal of pain (London, England) 2005;9(5):543–554. doi: 10.1016/j.ejpain.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Donaghue KC, Margan SH, Chan AK, Holloway B, Silink M, Rangel T, Bennetts B. The association of aldose reductase gene (AKR1B1) polymorphisms with diabetic neuropathy in adolescents. Diabet Med. 2005;22(10):1315–1320. doi: 10.1111/j.1464-5491.2005.01631.x. [DOI] [PubMed] [Google Scholar]
- Dorchy H. Lower plasma vitamin C levels in young type I diabetic patients with microalbuminuria. Journal of diabetes and its complications. 1999;13(2):119. doi: 10.1016/s1056-8727(99)00035-5. [DOI] [PubMed] [Google Scholar]
- Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. The Journal of clinical investigation. 2003;112(7):1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(22):12222–12226. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin RH, Jensen MP, Gammaitoni AR, Olaleye DO, Galer BS. Symptom profiles differ in patients with neuropathic versus non-neuropathic pain. J Pain. 2007a;8:118–126. doi: 10.1016/j.jpain.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Dworkin RH, O’Connor AB, Backonja M, Farrar JT, Finnerup NB, Jensen TS, Kalso EA, Loeser JD, Miaskowski C, Nurmikko TJ, Portenoy RK, Rice AS, Stacey BR, Treede RD, Turk DC, Wallace MS. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007b;132(3):237–251. doi: 10.1016/j.pain.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Dworkin RH, Turk DC, Farrar JT, Haythornthwaite JA, Jensen MP, Katz NP, Kerns RD, Stucki G, Allen RR, Bellamy N, Carr DB, Chandler J, Cowan P, Dionne R, Galer BS, Hertz S, Jadad AR, Kramer LD, Manning DC, Martin S, McCormick CG, McDermott MP, McGrath P, Quessy S, Rappaport BA, Robbins W, Robinson JP, Rothman M, Royal MA, Simon L, Stauffer JW, Stein W, Tollett J, Wernicke J, Witter J. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain. 2005;113:9–19. doi: 10.1016/j.pain.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Dyck PJ, Karnes JL, O’Brien PC, Litchy WJ, Low PA, Melton LJ., 3rd The Rochester Diabetic Neuropathy Study: reassessment of tests and criteria for diagnosis and staged severity. Neurology. 1992;42(6):1164–1170. doi: 10.1212/wnl.42.6.1164. [DOI] [PubMed] [Google Scholar]
- Dyck PJ, Kratz KM, Karnes JL, Litchy WJ, Klein R, Pach JM, Wilson DM, O’Brien PC, Melton LJ, 3rd, Service FJ. The prevalence by staged severity of various types of diabetic neuropathy, retinopathy, and nephropathy in a population-based cohort: the Rochester Diabetic Neuropathy Study. Neurology. 1993;43(4):817–824. doi: 10.1212/wnl.43.4.817. [DOI] [PubMed] [Google Scholar]
- Edelstein D, Brownlee M. Aminoguanidine ameliorates albuminuria in diabetic hypertensive rats. Diabetologia. 1992;35(1):96–97. doi: 10.1007/BF00400859. [DOI] [PubMed] [Google Scholar]
- Edwards AS, Faux MC, Scott JD, Newton AC. Carboxyl-terminal phosphorylation regulates the function and subcellular localization of protein kinase C betaII. The Journal of biological chemistry. 1999;274(10):6461–6468. doi: 10.1074/jbc.274.10.6461. [DOI] [PubMed] [Google Scholar]
- The effect of intensive diabetes therapy on the development and progression of neuropathy. The Diabetes Control and Complications Trial Research Group. Annals of internal medicine. 1995;122(8):561–568. doi: 10.7326/0003-4819-122-8-199504150-00001. [DOI] [PubMed] [Google Scholar]
- Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. Jama. 2002;287(19):2563–2569. doi: 10.1001/jama.287.19.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. The New England journal of medicine. 1993;329(14):977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- The effect of ruboxistaurin on visual loss in patients with moderately severe to very severe nonproliferative diabetic retinopathy: initial results of the Protein Kinase C beta Inhibitor Diabetic Retinopathy Study (PKC-DRS) multicenter randomized clinical trial. Diabetes. 2005;54(7):2188–2197. doi: 10.2337/diabetes.54.7.2188. [DOI] [PubMed] [Google Scholar]
- Eisenberg E, Lurie Y, Braker C, Daoud D, Ishay A. Lamotrigine reduces painful diabetic neuropathy: a randomized, controlled study. Neurology. 2001;57(3):505–509. doi: 10.1212/wnl.57.3.505. [DOI] [PubMed] [Google Scholar]
- Ekstrom AR, Kanje M, Skottner A. Nerve regeneration and serum levels of insulin-like growth factor-I in rats with streptozotocin-induced insulin deficiency. Brain research. 1989;496(1–2):141–147. doi: 10.1016/0006-8993(89)91060-3. [DOI] [PubMed] [Google Scholar]
- Elias KA, Cronin MJ, Stewart TA, Carlsen RC. Peripheral neuropathy in transgenic diabetic mice: restoration of C-fiber function with human recombinant nerve growth factor. Diabetes. 1998;47(10):1637–1642. doi: 10.2337/diabetes.47.10.1637. [DOI] [PubMed] [Google Scholar]
- England JD, Gronseth GS, Franklin G, Miller RG, Asbury AK, Carter GT, Cohen JA, Fisher MA, Howard JF, Kinsella LJ, Latov N, Lewis RA, Low PA, Sumner AJ. Distal symmetric polyneuropathy: a definition for clinical research: report of the American Academy of Neurology, the American Association of Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2005;64(2):199–207. doi: 10.1212/01.WNL.0000149522.32823.EA. [DOI] [PubMed] [Google Scholar]
- Eriksson JG, Forsen TJ, Mortensen SA, Rohde M. The effect of coenzyme Q10 administration on metabolic control in patients with type 2 diabetes mellitus. Bio Factors (Oxford, England) 1999;9(2–4):315–318. doi: 10.1002/biof.5520090229. [DOI] [PubMed] [Google Scholar]
- Fang X, Djouhri L, McMullan S, Berry C, Okuse K, Waxman SG, Lawson SN. trkA is expressed in nociceptive neurons and influences electrophysiological properties via Nav1.8 expression in rapidly conducting nociceptors. J Neurosci. 2005;25(19):4868–4878. doi: 10.1523/JNEUROSCI.0249-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman EL, SMJ, Greene DA. Diabetic neuropathy. In: Turtle KT JR, Osato S, editors. Diabetes in the New Millenium. Sydney: The Endocrinology and Diabetes Research Foundation of the University of Sydney; 1999. pp. 387–402. [Google Scholar]
- Feldman EL, Stevens MJ, Greene DA. Pathogenesis of diabetic neuropathy. Clinical neuroscience (New York, N Y. 1997;4(6):365–370. [PubMed] [Google Scholar]
- Feldman EL, Stevens MJ, Russell JW, Peltier A, Inzucchi S, Porte JD, Sherwin RS, Baron A. The Diabetes Mellitus Manual. Vol. 6. McGraw-Hill; 2005. Somatosensory neuropathy; pp. 366–384. [Google Scholar]
- Feldman EL, Stevens MJ, Russell JW, Sperling MA. Contemporary Endocrinology. Humana Press; 2002a. Diabetic peripheral and autonomic neuropathy. [Google Scholar]
- Feldman EL, Stevens MJ, Thomas PK, Brown MB, Canal N, Greene DA. A practical two-step quantitative clinical and electrophysiological assessment for the diagnosis and staging of diabetic neuropathy. Diabetes care. 1994;17(11):1281–1289. doi: 10.2337/diacare.17.11.1281. [DOI] [PubMed] [Google Scholar]
- Feldman EL, Stevens MJ, Russell JW, Greene DA. Somatosensory neuropathy. In: Porte SRSD Jr, Baron A, editors. Ellenberg and Rifkin’s Diabetes Mellitus. Philadelphia: McGraw Hill; 2002b. pp. 771–788. [Google Scholar]
- Fernandez S, Fernandez AM, Lopez-Lopez C, Torres-Aleman I. Emerging roles of insulin-like growth factor-I in the adult brain. Growth Horm IGF Res. 2007;17(2):89–95. doi: 10.1016/j.ghir.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Fernyhough P, Diemel LT, Brewster WJ, Tomlinson DR. Deficits in sciatic nerve neuropeptide content coincide with a reduction in target tissue nerve growth factor messenger RNA in streptozotocin-diabetic rats: effects of insulin treatment. Neuroscience. 1994;62(2):337–344. doi: 10.1016/0306-4522(94)90368-9. [DOI] [PubMed] [Google Scholar]
- Fernyhough P, Diemel LT, Brewster WJ, Tomlinson DR. Altered neurotrophin mRNA levels in peripheral nerve and skeletal muscle of experimentally diabetic rats. Journal of neurochemistry. 1995a;64(3):1231–1237. doi: 10.1046/j.1471-4159.1995.64031231.x. [DOI] [PubMed] [Google Scholar]
- Fernyhough P, Diemel LT, Hardy J, Brewster WJ, Mohiuddin L, Tomlinson DR. Human recombinant nerve growth factor replaces deficient neurotrophic support in the diabetic rat. The European journal of neuroscience. 1995b;7(5):1107–1110. doi: 10.1111/j.1460-9568.1995.tb01098.x. [DOI] [PubMed] [Google Scholar]
- Fernyhough P, Willars GB, Lindsay RM, Tomlinson DR. Insulin and insulin-like growth factor I enhance regeneration in cultured adult rat sensory neurones. Brain research. 1993;607(1–2):117–124. doi: 10.1016/0006-8993(93)91496-f. [DOI] [PubMed] [Google Scholar]
- Ford I, Cotter MA, Cameron NE, Greaves M. The effects of treatment with alpha-lipoic acid or evening primrose oil on vascular hemostatic and lipid risk factors, blood flow, and peripheral nerve conduction in the streptozotocin-diabetic rat. Metabolism: clinical and experimental. 2001;50(8):868–875. doi: 10.1053/meta.2001.24914. [DOI] [PubMed] [Google Scholar]
- Forrest KY, Maser RE, Pambianco G, Becker DJ, Orchard TJ. Hypertension as a risk factor for diabetic neuropathy: a prospective study. Diabetes. 1997;46(4):665–670. doi: 10.2337/diab.46.4.665. [DOI] [PubMed] [Google Scholar]
- Foster TS. Efficacy and safety of alpha-lipoic acid supplementation in the treatment of symptomatic diabetic neuropathy. The Diabetes educator. 2007;33(1):111–117. doi: 10.1177/0145721706297450. [DOI] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Developmental cell. 2001;1(4):515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Franklin GM, Kahn LB, Baxter J, Marshall JA, Hamman RF. Sensory neuropathy in non-insulin-dependent diabetes mellitus. The San Luis Valley Diabetes Study. American journal of epidemiology. 1990;131(4):633–643. doi: 10.1093/oxfordjournals.aje.a115547. [DOI] [PubMed] [Google Scholar]
- Freeman R. Autonomic peripheral neuropathy. Lancet. 2005;365(9466):1259–1270. doi: 10.1016/S0140-6736(05)74815-7. [DOI] [PubMed] [Google Scholar]
- Freeman R, Raskin P, Hewitt DJ, Vorsanger GJ, Jordan DM, Xiang J, Rosenthal NR. Randomized study of tramadol/acetaminophen versus placebo in painful diabetic peripheral neuropathy. Current medical research and opinion. 2007;23(1):147–161. doi: 10.1185/030079906X162674. [DOI] [PubMed] [Google Scholar]
- FreeStyle NavigatorTM. Continuous Glucose Monitoring System Use in Children with Type 1 Diabetes using glargine-based multiple daily dose regimens: Results of a Pilot Trial. (2007) Diabetes care. doi: 10.2337/dc07-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freynhagen R, Strojek K, Griesing T, Whalen E, Balkenohl M. Efficacy of pregabalin in neuropathic pain evaluated in a 12-week, randomised, double-blind, multicentre, placebo-controlled trial of flexible- and fixed-dose regimens. Pain. 2005;115(3):254–263. doi: 10.1016/j.pain.2005.02.032. [DOI] [PubMed] [Google Scholar]
- Fu MX, Thorpe SR, Baynes JW. Effects of aspirin on glycation, glycoxidation and cross-linking of collagen. In: Labuz TP, Reneccius GA, Monnier VM, OBrien JW, Baynes JW, editors. Maillard Reaction in Chemistry, Food, and Health. Cambridge: The Royal Society of Chemistry; 1994. pp. 95–100. [Google Scholar]
- Gabbay KH. Aldose reductase inhibition in the treatment of diabetic neuropathy: where are we in 2004? Current diabetes reports. 2004;4(6):405–408. doi: 10.1007/s11892-004-0047-z. [DOI] [PubMed] [Google Scholar]
- Gale EA, Bingley PJ, Emmett CL, Collier T. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004;363(9413):925–931. doi: 10.1016/S0140-6736(04)15786-3. [DOI] [PubMed] [Google Scholar]
- Garcia Soriano F, Virag L, Jagtap P, Szabo E, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ, Szabo C. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nature medicine. 2001;7(1):108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- Giannoukakis N. Fidarestat. Sanwa Kagaku/NC Curex/Sankyo. Curr Opin Investig Drugs. 2003;4(10):1233–1239. [PubMed] [Google Scholar]
- Gilron I, Bailey JM, Tu D, Holden RR, Weaver DF, Houlden RL. Morphine, gabapentin, or their combination for neuropathic pain. The New England journal of medicine. 2005a;352(13):1324–1334. doi: 10.1056/NEJMoa042580. [DOI] [PubMed] [Google Scholar]
- Gilron I, Max MB. Combination pharmacotherapy for neuropathic pain: current evidence and future directions. Expert review of neurotherapeutics. 2005b;5(6):823–830. doi: 10.1586/14737175.5.6.823. [DOI] [PubMed] [Google Scholar]
- Gimbel JS, Richards P, Portenoy RK. Controlled-release oxycodone for pain in diabetic neuropathy: a randomized controlled trial. Neurology. 2003;60(6):927–934. doi: 10.1212/01.wnl.0000057720.36503.2c. [DOI] [PubMed] [Google Scholar]
- Giugliano D, Acampora R, De Rosa N, Quatraro A, De Angelis L, Ceriello A, D’Onofrio F. Coronary artery disease in type-2 diabetes mellitus: a scintigraphic study. Diabete & metabolisme. 1993;19(5):463–466. [PubMed] [Google Scholar]
- Giuliano FA, Leriche A, Jaudinot EO, de Gendre AS. Prevalence of erectile dysfunction among 7689 patients with diabetes or hypertension, or both. Urology. 2004;64(6):1196–1201. doi: 10.1016/j.urology.2004.08.059. [DOI] [PubMed] [Google Scholar]
- Goldstein DJ, Lu Y, Detke MJ, Lee TC, Iyengar S. Duloxetine vs. placebo in patients with painful diabetic neuropathy. Pain. 2005;116:109–118. doi: 10.1016/j.pain.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Gomes MB, Piccirillo LJ, Nogueira VG, Matos HJ. Acute-phase proteins among patients with type 1 diabetes. Diabetes & metabolism. 2003;29(4 Pt 1):405–411. doi: 10.1016/s1262-3636(07)70051-4. [DOI] [PubMed] [Google Scholar]
- Gomez-Perez FJ, Choza R, Rios JM, Reza A, Huerta E, Aguilar CA, Rull JA. Nortriptyline-fluphenazine vs. carbamazepine in the symptomatic treatment of diabetic neuropathy. Archives of medical research. 1996;27(4):525–529. [PubMed] [Google Scholar]
- Gonzalez-Clemente JM, Mauricio D, Richart C, Broch M, Caixas A, Megia A, Gimenez-Palop O, Simon I, Martinez-Riquelme A, Gimenez-Perez G, Vendrell J. Diabetic neuropathy is associated with activation of the TNF-alpha system in subjects with type 1 diabetes mellitus. Clinical endocrinology. 2005;63(5):525–529. doi: 10.1111/j.1365-2265.2005.02376.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez ER, Oley MA. The management of lower-extremity diabetic ulcers. Managed care interface. 2000;13(11):80–87. [PubMed] [Google Scholar]
- Gordois A, Scuffham P, Shearer A, Oglesby A, Tobian JA. The health care costs of diabetic peripheral neuropathy in the US. Diabetes care. 2003;26(6):1790–1795. doi: 10.2337/diacare.26.6.1790. [DOI] [PubMed] [Google Scholar]
- Green K, Brand MD, Murphy MP. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes. 2004;53(Suppl 1):S110–118. doi: 10.2337/diabetes.53.2007.s110. [DOI] [PubMed] [Google Scholar]
- Greene DA, Arezzo JC, Brown MB. Effect of aldose reductase inhibition on nerve conduction and morphometry in diabetic neuropathy. Zenarestat Study Group. Neurology. 1999;53(3):580–591. doi: 10.1212/wnl.53.3.580. [DOI] [PubMed] [Google Scholar]
- Greene DA, Lattimer SA, Sima AA. Sorbitol, phosphoinositides, and sodium-potassium-ATPase in the pathogenesis of diabetic complications. The New England journal of medicine. 1987;316(10):599–606. doi: 10.1056/NEJM198703053161007. [DOI] [PubMed] [Google Scholar]
- Greene DA, Sima AA. Effects of aldose reductase inhibitors on the progression of nerve damage. Diabet Med. 1993;10(Suppl 2):31S–32S. doi: 10.1111/j.1464-5491.1993.tb00194.x. [DOI] [PubMed] [Google Scholar]
- Gruden G, Bruno G, Chaturvedi N, Burt D, Schalkwijk C, Pinach S, Stehouwer CD, Witte DR, Fuller JH, Cavallo Perin P. Serum Hsp27 and Diabetic Complications in the Eurodiab Prospective Complications Study: a Novel Circulating Marker for Diabetic Neuropathy. Diabetes. 2008 doi: 10.2337/db08-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumy LF, Bampton ET, Tolkovsky AM. Hyperglycaemia inhibits Schwann cell proliferation and migration and restricts regeneration of axons and Schwann cells from adult murine DRG. Molecular and cellular neurosciences. 2008;37(2):298–311. doi: 10.1016/j.mcn.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Ha HC, Hester LD, Snyder SH. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(5):3270–3275. doi: 10.1073/pnas.052712399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halat KM, Dennehy CE. Botanicals and dietary supplements in diabetic peripheral neuropathy. The Journal of the American Board of Family Practice / American Board of Family Practice. 2003;16(1):47–57. doi: 10.3122/jabfm.16.1.47. [DOI] [PubMed] [Google Scholar]
- Hammes HP, Du X, Edelstein D, Taguchi T, Matsumura T, Ju Q, Lin J, Bierhaus A, Nawroth P, Hannak D, Neumaier M, Bergfeld R, Giardino I, Brownlee M. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nature medicine. 2003;9(3):294–299. doi: 10.1038/nm834. [DOI] [PubMed] [Google Scholar]
- Hammes HP, Strodter D, Weiss A, Bretzel RG, Federlin K, Brownlee M. Secondary intervention with aminoguanidine retards the progression of diabetic retinopathy in the rat model. Diabetologia. 1995;38(6):656–660. doi: 10.1007/BF00401835. [DOI] [PubMed] [Google Scholar]
- Han HJ, Kang CW, Park SH. Tissue-specific regulation of insulin-like growth factors and insulin-like growth factor binding proteins in male diabetic rats in vivo and in vitro. Clinical and experimental pharmacology & physiology. 2006;33(12):1172–1179. doi: 10.1111/j.1440-1681.2006.04495.x. [DOI] [PubMed] [Google Scholar]
- Harati Y, Gooch C, Swenson M, Edelman S, Greene D, Raskin P, Donofrio P, Cornblath D, Sachdeo R, Siu CO, Kamin M. Double-blind randomized trial of tramadol for the treatment of the pain of diabetic neuropathy. Neurology. 1998;50:1842–1846. doi: 10.1212/wnl.50.6.1842. [DOI] [PubMed] [Google Scholar]
- Hasnis E, Bar-Shai M, Burbea Z, Reznick AZ. Mechanisms underlying cigarette smoke-induced NF-kappaB activation in human lymphocytes: the role of reactive nitrogen species. J Physiol Pharmacol. 2007;58(Suppl 5 Pt 1):275–287. [PubMed] [Google Scholar]
- Haupt E, Ledermann H, Kopcke W. Benfotiamine in the treatment of diabetic polyneuropathy--a three-week randomized, controlled pilot study (BEDIP study) International journal of clinical pharmacology and therapeutics. 2005;43(2):71–77. doi: 10.5414/cpp43071. [DOI] [PubMed] [Google Scholar]
- Hellweg R, Hartung HD. Endogenous levels of nerve growth factor (NGF) are altered in experimental diabetes mellitus: a possible role for NGF in the pathogenesis of diabetic neuropathy. Journal of neuroscience research. 1990;26(2):258–267. doi: 10.1002/jnr.490260217. [DOI] [PubMed] [Google Scholar]
- Hellweg R, Raivich G, Hartung HD, Hock C, Kreutzberg GW. Axonal transport of endogenous nerve growth factor (NGF) and NGF receptor in experimental diabetic neuropathy. Experimental neurology. 1994;130(1):24–30. doi: 10.1006/exnr.1994.1181. [DOI] [PubMed] [Google Scholar]
- Hellweg R, Wohrle M, Hartung HD, Stracke H, Hock C, Federlin K. Diabetes mellitus-associated decrease in nerve growth factor levels is reversed by allogeneic pancreatic islet transplantation. Neuroscience letters. 1991;125(1):1–4. doi: 10.1016/0304-3940(91)90114-9. [DOI] [PubMed] [Google Scholar]
- Hennekens CH, Knatterud GL, Pfeffer MA. Use of aspirin to reduce risks of cardiovascular disease in patients with diabetes: clinical and research challenges. Diabetes care. 2004;27(11):2752–2754. doi: 10.2337/diacare.27.11.2752. [DOI] [PubMed] [Google Scholar]
- Hers HG. The mechanism of the transformation of glucose in fructose in the seminal vesicles. Biochimica et biophysica acta. 1956;22(1):202–203. doi: 10.1016/0006-3002(56)90247-5. [DOI] [PubMed] [Google Scholar]
- Hirakata Y, Kitamura S. Elevated serum transforming growth factor beta 1 level in primary lung cancer patients with finger clubbing. European journal of clinical investigation. 1996;26(9):820–823. doi: 10.1046/j.1365-2362.1996.2260560.x. [DOI] [PubMed] [Google Scholar]
- Hirsch IB, Brownlee M. Should minimal blood glucose variability become the gold standard of glycemic control? Journal of diabetes and its complications. 2005;19(3):178–181. doi: 10.1016/j.jdiacomp.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Hogan P, Dall T, Nikolov P. Economic costs of diabetes in the US in 2002. Diabetes care. 2003;26(3):917–932. doi: 10.2337/diacare.26.3.917. [DOI] [PubMed] [Google Scholar]
- Hotta N, Akanuma Y, Kawamori R, Matsuoka K, Oka Y, Shichiri M, Toyota T, Nakashima M, Yoshimura I, Sakamoto N, Shigeta Y. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: the 3-year, multicenter, comparative Aldose Reductase Inhibitor-Diabetes Complications Trial. Diabetes care. 2006;29(7):1538–1544. doi: 10.2337/dc05-2370. [DOI] [PubMed] [Google Scholar]
- Hounsom L, Horrobin DF, Tritschler H, Corder R, Tomlinson DR. A lipoic acid-gamma linolenic acid conjugate is effective against multiple indices of experimental diabetic neuropathy. Diabetologia. 1998;41(7):839–843. doi: 10.1007/s001250050996. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annual review of biochemistry. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Huang TJ, Verkhratsky A, Fernyhough P. Insulin enhances mitochondrial inner membrane potential and increases ATP levels through phosphoinositide 3-kinase in adult sensory neurons. Molecular and cellular neurosciences. 2005;28(1):42–54. doi: 10.1016/j.mcn.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Hudson BI, Schmidt AM. RAGE: a novel target for drug intervention in diabetic vascular disease. Pharmaceutical research. 2004;21(7):1079–1086. doi: 10.1023/b:pham.0000032992.75423.9b. [DOI] [PubMed] [Google Scholar]
- Ilnytska O, Lyzogubov VV, Stevens MJ, Drel VR, Mashtalir N, Pacher P, Yorek MA, Obrosova IG. Poly(ADP-ribose) polymerase inhibition alleviates experimental diabetic sensory neuropathy. Diabetes. 2006;55(6):1686–1694. doi: 10.2337/db06-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352(9131):837–853. [PubMed] [Google Scholar]
- Ishii DN, Lupien SB. Insulin-like growth factors protect against diabetic neuropathy: effects on sensory nerve regeneration in rats. Journal of neuroscience research. 1995;40(1):138–144. doi: 10.1002/jnr.490400116. [DOI] [PubMed] [Google Scholar]
- Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A, Bursell SE, Kern TS, Ballas LM, Heath WF, Stramm LE, Feener EP, King GL. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science (New York, N Y. 1996;272(5262):728–731. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- Itoh K, Ishii T, Wakabayashi N, Yamamoto M. Regulatory mechanisms of cellular response to oxidative stress. Free radical research. 1999;31(4):319–324. doi: 10.1080/10715769900300881. [DOI] [PubMed] [Google Scholar]
- Jacobs EJ, Henion AK, Briggs PJ, Connell CJ, McCullough ML, Jonas CR, Rodriguez C, Calle EE, Thun MJ. Vitamin C and vitamin E supplement use and bladder cancer mortality in a large cohort of US men and women. American journal of epidemiology. 2002;156(11):1002–1010. doi: 10.1093/aje/kwf147. [DOI] [PubMed] [Google Scholar]
- Jann MW, Slade JH. Antidepressant agents for the treatment of chronic pain and depression. Pharmacotherapy. 2007;27(11):1571–1587. doi: 10.1592/phco.27.11.1571. [DOI] [PubMed] [Google Scholar]
- Jarvis B, Coukell AJ. Mexiletine. A review of its therapeutic use in painful diabetic neuropathy. Drugs. 1998;56(4):691–707. doi: 10.2165/00003495-199856040-00016. [DOI] [PubMed] [Google Scholar]
- Je HD, Shin CY, Park SY, Yim SH, Kum C, Huh IH, Kim JH, Sohn UD. Combination of vitamin C and rutin on neuropathy and lung damage of diabetes mellitus rats. Archives of pharmacal research. 2002;25(2):184–190. doi: 10.1007/BF02976561. [DOI] [PubMed] [Google Scholar]
- Jensen MP, Dworkin RH, Gammaitoni AR, Olaleye DO, Oleka N, Galer BS. Do pain qualities and spatial characteristics make independent contributions to interference with physical and emotional functioning? J Pain. 2006a;7:644–653. doi: 10.1016/j.jpain.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Jensen TS, Backonja MM, Hernandez Jimenez S, Tesfaye S, Valensi P, Ziegler D. New perspectives on the management of diabetic peripheral neuropathic pain. Diab Vasc Dis Res. 2006b;3(2):108–119. doi: 10.3132/dvdr.2006.013. [DOI] [PubMed] [Google Scholar]
- Jermendy G, Toth L, Voros P, Koltai MZ, Pogatsa G. Cardiac autonomic neuropathy and QT interval length. A follow-up study in diabetic patients. Acta cardiologica. 1991;46(2):189–200. [PubMed] [Google Scholar]
- Judzewitsch RG, Jaspan JB, Polonsky KS, Weinberg CR, Halter JB, Halar E, Pfeifer MA, Vukadinovic C, Bernstein L, Schneider M, Liang KY, Gabbay KH, Rubenstein AH, Porte D., Jr Aldose reductase inhibition improves nerve conduction velocity in diabetic patients. The New England journal of medicine. 1983;308(3):119–125. doi: 10.1056/NEJM198301203080302. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Murakawa Y, Zhang W, Sima AA. Unmyelinated fiber sensory neuropathy differs in type 1 and type 2 diabetes. Diabetes/metabolism research and reviews. 2005;21(5):448–458. doi: 10.1002/dmrr.541. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Zhang W, Ekberg K, Wahren J, Sima AA. C-Peptide reverses nociceptive neuropathy in type 1 diabetes. Diabetes. 2006;55(12):3581–3587. doi: 10.2337/db06-0396. [DOI] [PubMed] [Google Scholar]
- Kaneto H, Xu G, Song KH, Suzuma K, Bonner-Weir S, Sharma A, Weir GC. Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function through the induction of oxidative stress. The Journal of biological chemistry. 2001;276(33):31099–31104. doi: 10.1074/jbc.M104115200. [DOI] [PubMed] [Google Scholar]
- Kang KW, Lee SJ, Kim SG. Molecular mechanism of nrf2 activation by oxidative stress. Antioxidants & redox signaling. 2005;7(11–12):1664–1673. doi: 10.1089/ars.2005.7.1664. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Current opinion in neurobiology. 2000;10(3):381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kasayama S, Oka T. Impaired production of nerve growth factor in the submandibular gland of diabetic mice. The American journal of physiology. 1989;257(3 Pt 1):E400–404. doi: 10.1152/ajpendo.1989.257.3.E400. [DOI] [PubMed] [Google Scholar]
- Kato N, Mizuno K, Makino M, Suzuki T, Yagihashi S. Effects of 15-month aldose reductase inhibition with fidarestat on the experimental diabetic neuropathy in rats. Diabetes research and clinical practice. 2000;50(2):77–85. doi: 10.1016/s0168-8227(00)00164-9. [DOI] [PubMed] [Google Scholar]
- Kawamura N, Dyck PJ, Schmeichel AM, Engelstad JK, Low PA, Dyck PJ. Inflammatory mediators in diabetic and non-diabetic lumbosacral radiculoplexus neuropathy. Acta neuropathologica. 2008;115(2):231–239. doi: 10.1007/s00401-007-0326-2. [DOI] [PubMed] [Google Scholar]
- Kellogg AP, Pop-Busui R. Peripheral nerve dysfunction in experimental diabetes is mediated by cyclooxygenase-2 and oxidative stress. Antioxidants & redox signaling. 2005;7(11–12):1521–1529. doi: 10.1089/ars.2005.7.1521. [DOI] [PubMed] [Google Scholar]
- Kellogg AP, Wiggin TD, Larkin DD, Hayes JM, Stevens MJ, Pop-Busui R. Protective effects of cyclooxygenase-2 gene inactivation against peripheral nerve dysfunction and intraepidermal nerve fiber loss in experimental diabetes. Diabetes. 2007;56(12):2997–3005. doi: 10.2337/db07-0740. [DOI] [PubMed] [Google Scholar]
- Kennedy WR, Navarro X, Sutherland DE. Neuropathy profile of diabetic patients in a pancreas transplantation program. Neurology. 1995;45(4):773–780. doi: 10.1212/wnl.45.4.773. [DOI] [PubMed] [Google Scholar]
- Kim YW, Zhao RJ, Park SJ, Lee JR, Cho IJ, Yang CH, Kim SG, Kim SC. Anti-inflammatory effects of liquiritigenin as a consequence of the inhibition of NF-kappaB-dependent iNOS and proinflammatory cytokines production. British journal of pharmacology. 2008 doi: 10.1038/bjp.2008.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita JH, Dvornik D, Kraml M, Gabbay KH. The effect of an aldose reductase inhibitor on the galactose-exposed rabbit lens. Biochimica et biophysica acta. 1968;158(3):472–475. doi: 10.1016/0304-4165(68)90305-x. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Mizisin AP. CNTFR alpha alone or in combination with CNTF promotes macrophage chemotaxis in vitro. Neuropeptides. 2000;34(6):338–347. doi: 10.1054/npep.2000.0829. [DOI] [PubMed] [Google Scholar]
- Kochar DK, Rawat N, Agrawal RP, Vyas A, Beniwal R, Kochar SK, Garg P. Sodium valproate for painful diabetic neuropathy: a randomized double-blind placebo-controlled study. Qjm. 2004;97(1):33–38. doi: 10.1093/qjmed/hch007. [DOI] [PubMed] [Google Scholar]
- Kolm-Litty V, Sauer U, Nerlich A, Lehmann R, Schleicher ED. High glucose-induced transforming growth factor beta1 production is mediated by the hexosamine pathway in porcine glomerular mesangial cells. The Journal of clinical investigation. 1998;101(1):160–169. doi: 10.1172/JCI119875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong MF, Horowitz M, Jones KL, Wishart JM, Harding PE. Natural history of diabetic gastroparesis. Diabetes care. 1999;22(3):503–507. doi: 10.2337/diacare.22.3.503. [DOI] [PubMed] [Google Scholar]
- Kumar A, Kaundal RK, Iyer S, Sharma SS. Effects of resveratrol on nerve functions, oxidative stress and DNA fragmentation in experimental diabetic neuropathy. Life sciences. 2007;80(13):1236–1244. doi: 10.1016/j.lfs.2006.12.036. [DOI] [PubMed] [Google Scholar]
- Kumar D, Marshall HJ. Diabetic peripheral neuropathy: amelioration of pain with transcutaneous electrostimulation. Diabetes care. 1997;20(11):1702–1705. doi: 10.2337/diacare.20.11.1702. [DOI] [PubMed] [Google Scholar]
- Labinskyy N, Csiszar A, Veress G, Stef G, Pacher P, Oroszi G, Wu J, Ungvari Z. Vascular dysfunction in aging: potential effects of resveratrol, an anti-inflammatory phytoestrogen. Current medicinal chemistry. 2006;13(9):989–996. doi: 10.2174/092986706776360987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KM, Kang BS, Lee HL, Son SJ, Hwang SH, Kim DS, Park JS, Cho HJ. Spinal NF-kB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity. The European journal of neuroscience. 2004a;19(12):3375–3381. doi: 10.1111/j.0953-816X.2004.03441.x. [DOI] [PubMed] [Google Scholar]
- Lee TC, Barshes NR, O’Mahony CA, Nguyen L, Brunicardi FC, Ricordi C, Alejandro R, Schock AP, Mote A, Goss JA. The effect of pancreatic islet transplantation on progression of diabetic retinopathy and neuropathy. Transplantation proceedings. 2005;37(5):2263–2265. doi: 10.1016/j.transproceed.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Molecular biology of the cell. 2004b;15(11):5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinninger GM, Backus C, Sastry AM, Yi YB, Wang CW, Feldman EL. Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiology of disease. 2006a;23(1):11–22. doi: 10.1016/j.nbd.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Edwards JL, Lipshaw MJ, Feldman EL. Mechanisms of disease: mitochondria as new therapeutic targets in diabetic neuropathy. Nature clinical practice. 2006b;2(11):620–628. doi: 10.1038/ncpneuro0320. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Feldman EL. Insulin-like growth factors in the treatment of neurological disease. Endocrine development. 2005;9:135–159. doi: 10.1159/000085763. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Vincent AM, Feldman EL. The role of growth factors in diabetic peripheral neuropathy. J Peripher Nerv Syst. 2004;9(1):26–53. doi: 10.1111/j.1085-9489.2004.09105.x. [DOI] [PubMed] [Google Scholar]
- Lesser H, Sharma U, LaMoreaux L, Poole RM. Pregabalin relieves symptoms of painful diabetic neuropathy: a randomized controlled trial. Neurology. 2004;63(11):2104–2110. doi: 10.1212/01.wnl.0000145767.36287.a1. [DOI] [PubMed] [Google Scholar]
- Levy D, Zochodne DW. NO pain: potential roles of nitric oxide in neuropathic pain. Pain Pract. 2004;4(1):11–18. doi: 10.1111/j.1533-2500.2004.04002.x. [DOI] [PubMed] [Google Scholar]
- Lewis MA, Hunihan L, Franco D, Robertson B, Palmer J, Laurent DR, Balasubramanian BN, Li Y, Westphal RS. Identification and characterization of compounds that potentiate NT-3-mediated Trk receptor activity. Molecular pharmacology. 2006;69(4):1396–1404. doi: 10.1124/mol.105.020255. [DOI] [PubMed] [Google Scholar]
- Li F, Abatan OI, Kim H, Burnett D, Larkin D, Obrosova IG, Stevens MJ. Taurine reverses neurological and neurovascular deficits in Zucker diabetic fatty rats. Neurobiology of disease. 2006;22(3):669–676. doi: 10.1016/j.nbd.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Li F, Drel VR, Szabo C, Stevens MJ, Obrosova IG. Low-dose poly(ADP-ribose) polymerase inhibitor-containing combination therapies reverse early peripheral diabetic neuropathy. Diabetes. 2005;54(5):1514–1522. doi: 10.2337/diabetes.54.5.1514. [DOI] [PubMed] [Google Scholar]
- Lindsay RM. Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J Neurosci. 1988;8(7):2394–2405. doi: 10.1523/JNEUROSCI.08-07-02394.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay RM. Role of neurotrophins and trk receptors in the development and maintenance of sensory neurons: an overview. Philosophical transactions of the Royal Society of London. 1996;351(1338):365–373. doi: 10.1098/rstb.1996.0030. [DOI] [PubMed] [Google Scholar]
- Little AA, Edwards JL, Feldman EL. Diabetic neuropathies. Practical neurology. 2007;7(2):82–92. [PubMed] [Google Scholar]
- Little WC, Zile MR, Kitzman DW, Hundley WG, O’Brien TX, Degroof RC. The effect of alagebrium chloride (ALT-711), a novel glucose cross-link breaker, in the treatment of elderly patients with diastolic heart failure. Journal of cardiac failure. 2005;11(3):191–195. doi: 10.1016/j.cardfail.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Low PA, Benrud-Larson LM, Sletten DM, Opfer-Gehrking TL, Weigand SD, O’Brien PC, Suarez GA, Dyck PJ. Autonomic symptoms and diabetic neuropathy: a population-based study. Diabetes care. 2004;27(12):2942–2947. doi: 10.2337/diacare.27.12.2942. [DOI] [PubMed] [Google Scholar]
- Low PA, Zimmerman BR, Dyck PJ. Comparison of distal sympathetic with vagal function in diabetic neuropathy. Muscle & nerve. 1986;9(7):592–596. doi: 10.1002/mus.880090703. [DOI] [PubMed] [Google Scholar]
- Lupien SB, Bluhm EJ, Ishii DN. Systemic insulin-like growth factor-I administration prevents cognitive impairment in diabetic rats, and brain IGF regulates learning/memory in normal adult rats. Journal of neuroscience research. 2003;74(4):512–523. doi: 10.1002/jnr.10791. [DOI] [PubMed] [Google Scholar]
- Makita Z, Vlassara H, Rayfield E, Cartwright K, Friedman E, Rodby R, Cerami A, Bucala R. Hemoglobin-AGE: a circulating marker of advanced glycosylation. Science (New York, N Y. 1992;258(5082):651–653. doi: 10.1126/science.1411574. [DOI] [PubMed] [Google Scholar]
- Malik RA. Can diabetic neuropathy be prevented by angiotensin-converting enzyme inhibitors? Annals of medicine. 2000;32(1):1–5. doi: 10.3109/07853890008995903. [DOI] [PubMed] [Google Scholar]
- Manzella D, Barbieri M, Ragno E, Paolisso G. Chronic administration of pharmacologic doses of vitamin E improves the cardiac autonomic nervous system in patients with type 2 diabetes. The American journal of clinical nutrition. 2001;73(6):1052–1057. doi: 10.1093/ajcn/73.6.1052. [DOI] [PubMed] [Google Scholar]
- Martin CL, Albers J, Herman WH, Cleary P, Waberski B, Greene DA, Stevens MJ, Feldman EL. Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes care. 2006;29(2):340–344. doi: 10.2337/diacare.29.02.06.dc05-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maser RE, Mitchell BD, Vinik AI, Freeman R. The association between cardiovascular autonomic neuropathy and mortality in individuals with diabetes: a meta-analysis. Diabetes care. 2003;26(6):1895–1901. doi: 10.2337/diacare.26.6.1895. [DOI] [PubMed] [Google Scholar]
- Maser RE, Pfeifer MA, Dorman JS, Kuller LH, Becker DJ, Orchard TJ. Diabetic autonomic neuropathy and cardiovascular risk. Pittsburgh Epidemiology of Diabetes Complications Study III. Archives of internal medicine. 1990;150(6):1218–1222. [PubMed] [Google Scholar]
- Matsunaga A, Kawamoto M, Shiraishi S, Yasuda T, Kajiyama S, Kurita S, Yuge O. Intrathecally administered COX-2 but not COX-1 or COX-3 inhibitors attenuate streptozotocin-induced mechanical hyperalgesia in rats. European journal of pharmacology. 2007;554(1):12–17. doi: 10.1016/j.ejphar.2006.09.072. [DOI] [PubMed] [Google Scholar]
- Max MB, Culnane M, Schafer SC, Gracely RH, Walther DJ, Smoller B, Dubner R. Amitriptyline relieves diabetic neuropathy pain in patients with normal or depressed mood. Neurology. 1987;37(4):589–596. doi: 10.1212/wnl.37.4.589. [DOI] [PubMed] [Google Scholar]
- Max MB, Lynch SA, Muir J, Shoaf SE, Smoller B, Dubner R. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. New England Journal of Medicine. 1992;326:1250–1256. doi: 10.1056/NEJM199205073261904. [DOI] [PubMed] [Google Scholar]
- McDonald DS, Cheng C, Martinez JA, Zochodne DW. Regenerative arrest of inflamed peripheral nerves: role of nitric oxide. Neuroreport. 2007;18(16):1635–1640. doi: 10.1097/WNR.0b013e3282f03fff. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Armanini MP, Ling LH, Phillips HS. Expression and coexpression of Trk receptors in subpopulations of adult primary sensory neurons projecting to identified peripheral targets. Neuron. 1994;12(5):1161–1171. doi: 10.1016/0896-6273(94)90323-9. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Bennett DL, Priestley JV, Shelton DL. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nature medicine. 1995;1(8):774–780. doi: 10.1038/nm0895-774. [DOI] [PubMed] [Google Scholar]
- Migdalis IN, Kalogeropoulou K, Kalantzis L, Nounopoulos C, Bouloukos A, Samartzis M. Insulin-like growth factor-I and IGF-I receptors in diabetic patients with neuropathy. Diabet Med. 1995;12(9):823–827. doi: 10.1111/j.1464-5491.1995.tb02086.x. [DOI] [PubMed] [Google Scholar]
- Miyauchi Y, Shikama H, Takasu T, Okamiya H, Umeda M, Hirasaki E, Ohhata I, Nakayama H, Nakagawa S. Slowing of peripheral motor nerve conduction was ameliorated by aminoguanidine in streptozocin-induced diabetic rats. European journal of endocrinology / European Federation of Endocrine Societies. 1996;134(4):467–473. doi: 10.1530/eje.0.1340467. [DOI] [PubMed] [Google Scholar]
- Mizisin AP, Bache M, DiStefano PS, Acheson A, Lindsay RM, Calcutt NA. BDNF attenuates functional and structural disorders in nerves of galactose-fed rats. Journal of neuropathology and experimental neurology. 1997;56(12):1290–1301. doi: 10.1097/00005072-199712000-00004. [DOI] [PubMed] [Google Scholar]
- Mizisin AP, DiStefano PS, Liu X, Garrett DN, Tonra JR. Decreased accumulation of endogenous brain-derived neurotrophic factor against constricting sciatic nerve ligatures in streptozotocin-diabetic and galactose-fed rats. Neuroscience letters. 1999;263(2–3):149–152. doi: 10.1016/s0304-3940(99)00131-7. [DOI] [PubMed] [Google Scholar]
- Mizisin AP, Kalichman MW, Bache M, Dines KC, DiStefano PS. NT-3 attenuates functional and structural disorders in sensory nerves of galactose-fed rats. Journal of neuropathology and experimental neurology. 1998;57(9):803–813. doi: 10.1097/00005072-199809000-00001. [DOI] [PubMed] [Google Scholar]
- Mizisin AP, Vu Y, Shuff M, Calcutt NA. Ciliary neurotrophic factor improves nerve conduction and ameliorates regeneration deficits in diabetic rats. Diabetes. 2004;53(7):1807–1812. doi: 10.2337/diabetes.53.7.1807. [DOI] [PubMed] [Google Scholar]
- Monnier L, Mas E, Ginet C, Michel F, Villon L, Cristol JP, Colette C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. Jama. 2006;295(14):1681–1687. doi: 10.1001/jama.295.14.1681. [DOI] [PubMed] [Google Scholar]
- Morello CM, Leckband SG, Stoner CP, Moorhouse DF, Sahagian GA. Randomized double-blind study comparing the efficacy of gabapentin with amitriptyline on diabetic peripheral neuropathy pain. Archives of internal medicine. 1999;159(16):1931–1937. doi: 10.1001/archinte.159.16.1931. [DOI] [PubMed] [Google Scholar]
- Nakamura J, Kato K, Hamada Y, Nakayama M, Chaya S, Nakashima E, Naruse K, Kasuya Y, Mizubayashi R, Miwa K, Yasuda Y, Kamiya H, Ienaga K, Sakakibara F, Koh N, Hotta N. A protein kinase C-beta-selective inhibitor ameliorates neural dysfunction in streptozotocin-induced diabetic rats. Diabetes. 1999;48(10):2090–2095. doi: 10.2337/diabetes.48.10.2090. [DOI] [PubMed] [Google Scholar]
- Nangle MR, Gibson TM, Cotter MA, Cameron NE. Effects of eugenol on nerve and vascular dysfunction in streptozotocin-diabetic rats. Planta medica. 2006;72(6):494–500. doi: 10.1055/s-2005-916262. [DOI] [PubMed] [Google Scholar]
- Naruse K, Rask-Madsen C, Takahara N, Ha SW, Suzuma K, Way KJ, Jacobs JR, Clermont AC, Ueki K, Ohshiro Y, Zhang J, Goldfine AB, King GL. Activation of vascular protein kinase C-beta inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes. 2006;55(3):691–698. doi: 10.2337/diabetes.55.03.06.db05-0771. [DOI] [PubMed] [Google Scholar]
- Navarro X, Sutherland DE, Kennedy WR. Long-term effects of pancreatic transplantation on diabetic neuropathy. Annals of neurology. 1997;42(5):727–736. doi: 10.1002/ana.410420509. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- O’Brien IA, Lewin IG, O’Hare JP, Arendt J, Corrall RJ. Abnormal circadian rhythm of melatonin in diabetic autonomic neuropathy. Clinical endocrinology. 1986;24(4):359–364. doi: 10.1111/j.1365-2265.1986.tb01639.x. [DOI] [PubMed] [Google Scholar]
- Oates PJ. Aldose Reductase, Still a Compelling Target for Diabetic Neuropathy. Current drug targets. 2008 doi: 10.2174/138945008783431781. in press. [DOI] [PubMed] [Google Scholar]
- Obrosova IG, Drel VR, Oltman CL, Mashtalir N, Tibrewala J, Groves JT, Yorek MA. Role of nitrosative stress in early neuropathy and vascular dysfunction in streptozotocin-diabetic rats. American journal of physiology. 2007;293(6):E1645–1655. doi: 10.1152/ajpendo.00479.2007. [DOI] [PubMed] [Google Scholar]
- Obrosova IG, Drel VR, Pacher P, Ilnytska O, Wang ZQ, Stevens MJ, Yorek MA. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes. 2005a;54(12):3435–3441. doi: 10.2337/diabetes.54.12.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrosova IG, Julius UA. Role for poly(ADP-ribose) polymerase activation in diabetic nephropathy, neuropathy and retinopathy. Current vascular pharmacology. 2005b;3(3):267–283. doi: 10.2174/1570161054368634. [DOI] [PubMed] [Google Scholar]
- Obrosova IG, Li F, Abatan OI, Forsell MA, Komjati K, Pacher P, Szabo C, Stevens MJ. Role of poly(ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes. 2004;53(3):711–720. doi: 10.2337/diabetes.53.3.711. [DOI] [PubMed] [Google Scholar]
- Obrosova IG, Mabley JG, Zsengeller Z, Charniauskaya T, Abatan OI, Groves JT, Szabo C. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. Faseb J. 2005c;19(3):401–403. doi: 10.1096/fj.04-1913fje. [DOI] [PubMed] [Google Scholar]
- Obrosova IG, Van Huysen C, Fathallah L, Cao XC, Greene DA, Stevens MJ. An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. Faseb J. 2002;16(1):123–125. doi: 10.1096/fj.01-0603fje. [DOI] [PubMed] [Google Scholar]
- Olchovsky D, Bruno JF, Gelato MC, Song J, Berelowitz M. Pituitary insulin-like growth factor-I content and gene expression in the streptozotocin-diabetic rat: evidence for tissue-specific regulation. Endocrinology. 1991;128(2):923–928. doi: 10.1210/endo-128-2-923. [DOI] [PubMed] [Google Scholar]
- Osawa T, Kato Y. Protective role of antioxidative food factors in oxidative stress caused by hyperglycemia. Annals of the New York Academy of Sciences. 2005;1043:440–451. doi: 10.1196/annals.1333.050. [DOI] [PubMed] [Google Scholar]
- Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51(2):514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- Packer L, Tritschler HJ, Wessel K. Neuroprotection by the metabolic antioxidant alpha-lipoic acid. Free radical biology & medicine. 1997;22(1–2):359–378. doi: 10.1016/s0891-5849(96)00269-9. [DOI] [PubMed] [Google Scholar]
- Palumbo PJ, Elveback LR, Whisnant JP. Neurologic complications of diabetes mellitus: transient ischemic attack, stroke, and peripheral neuropathy. Adv Neurol. 1978;19:593–601. [PubMed] [Google Scholar]
- Partanen J, Niskanen L, Lehtinen J, Mervaala E, Siitonen O, Uusitupa M. Natural history of peripheral neuropathy in patients with non-insulin-dependent diabetes mellitus. The New England journal of medicine. 1995;333(2):89–94. doi: 10.1056/NEJM199507133330203. [DOI] [PubMed] [Google Scholar]
- Peppa M, Brem H, Cai W, Zhang JG, Basgen J, Li Z, Vlassara H, Uribarri J. Prevention and reversal of diabetic nephropathy in db/db mice treated with alagebrium (ALT-711) American journal of nephrology. 2006;26(5):430–436. doi: 10.1159/000095786. [DOI] [PubMed] [Google Scholar]
- Perry E. Commentary: botanical potentials in Alzheimer’s disease. Journal of alternative and complementary medicine (New York, N Y. 2007;13(3):345–346. doi: 10.1089/acm.2006.6405. [DOI] [PubMed] [Google Scholar]
- Pertovaara A, Wei H, Kalmari J, Ruotsalainen M. Pain behavior and response properties of spinal dorsal horn neurons following experimental diabetic neuropathy in the rat: modulation by nitecapone, a COMT inhibitor with antioxidant properties. Experimental neurology. 2001;167(2):425–434. doi: 10.1006/exnr.2000.7574. [DOI] [PubMed] [Google Scholar]
- Pfeifer MA, Weinberg CR, Cook DL, Reenan A, Halter JB, Ensinck JW, Porte D., Jr Autonomic neural dysfunction in recently diagnosed diabetic subjects. Diabetes care. 1984;7(5):447–453. doi: 10.2337/diacare.7.5.447. [DOI] [PubMed] [Google Scholar]
- Pierson CR, Zhang W, Murakawa Y, Sima AA. Insulin deficiency rather than hyperglycemia accounts for impaired neurotrophic responses and nerve fiber regeneration in type 1 diabetic neuropathy. Journal of neuropathology and experimental neurology. 2003;62(3):260–271. doi: 10.1093/jnen/62.3.260. [DOI] [PubMed] [Google Scholar]
- Pirart J. Diabetes mellitus and its degenerative complications: a prospective study of 4,400 patients observed between 1947 and 1973. Diabetes care. 1978;1(3):168–188. [Google Scholar]
- Podar T, Tuomilehto J. The role of angiotensin converting enzyme inhibitors and angiotensin II receptor antagonists in the management of diabetic complications. Drugs. 2002;62(14):2007–2012. doi: 10.2165/00003495-200262140-00001. [DOI] [PubMed] [Google Scholar]
- Pop-Busui R, Sullivan KA, Van Huysen C, Bayer L, Cao X, Towns R, Stevens MJ. Depletion of taurine in experimental diabetic neuropathy: implications for nerve metabolic, vascular, and functional deficits. Experimental neurology. 2001;168(2):259–272. doi: 10.1006/exnr.2000.7591. [DOI] [PubMed] [Google Scholar]
- Price SA, Zeef LA, Wardleworth L, Hayes A, Tomlinson DR. Identification of changes in gene expression in dorsal root ganglia in diabetic neuropathy: correlation with functional deficits. Journal of neuropathology and experimental neurology. 2006;65(7):722–732. doi: 10.1097/01.jnen.0000228199.89420.90. [DOI] [PubMed] [Google Scholar]
- Prince PS, Menon VP. Antioxidant activity of Tinospora cordifolia roots in experimental diabetes. Journal of ethnopharmacology. 1999;65(3):277–281. doi: 10.1016/s0378-8741(98)00164-0. [DOI] [PubMed] [Google Scholar]
- Qiang X, Satoh J, Sagara M, Fukuzawa M, Masuda T, Sakata Y, Muto G, Muto Y, Takahashi K, Toyota T. Inhibitory effect of troglitazone on diabetic neuropathy in streptozotocin-induced diabetic rats. Diabetologia. 1998;41(11):1321–1326. doi: 10.1007/s001250051072. [DOI] [PubMed] [Google Scholar]
- Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. 2005;15(7):16R–28R. doi: 10.1093/glycob/cwi053. [DOI] [PubMed] [Google Scholar]
- Ramasamy R, Yan SF, Schmidt AM. Arguing for the motion: yes, RAGE is a receptor for advanced glycation endproducts. Molecular nutrition & food research. 2007;51(9):1111–1115. doi: 10.1002/mnfr.200700008. [DOI] [PubMed] [Google Scholar]
- Ramsey SD, Newton K, Blough D, McCulloch DK, Sandhu N, Reiber GE, Wagner EH. Incidence, outcomes, and cost of foot ulcers in patients with diabetes. Diabetes care. 1999;22(3):382–387. doi: 10.2337/diacare.22.3.382. [DOI] [PubMed] [Google Scholar]
- Raskin J, Pritchett YL, Wang F, D’Souza DN, Waninger AL, Iyengar S, Wernicke JF. A double-blind, randomized multicenter trial comparing duloxetine with placebo in the management of diabetic peripheral neuropathic pain. Pain medicine (Malden, Mass. 2005;6(5):346–356. doi: 10.1111/j.1526-4637.2005.00061.x. [DOI] [PubMed] [Google Scholar]
- Raskin P, Donofrio PD, Rosenthal NR, Hewitt DJ, Jordan DM, Xiang J, Vinik AI. Topiramate vs placebo in painful diabetic neuropathy: analgesic and metabolic effects. Neurology. 2004;63(5):865–873. doi: 10.1212/01.wnl.0000137341.89781.14. [DOI] [PubMed] [Google Scholar]
- Recio-Pinto E, Rechler MM, Ishii DN. Effects of insulin, insulin-like growth factor-II, and nerve growth factor on neurite formation and survival in cultured sympathetic and sensory neurons. J Neurosci. 1986;6(5):1211–1219. doi: 10.1523/JNEUROSCI.06-05-01211.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relative accuracy of the BD Logic and FreeStyle blood glucose meters. Diabetes technology & therapeutics. 2007;9(2):165–168. doi: 10.1089/dia.2006.0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter RW, Portenoy R, Sharma U, Lamoreaux L, Bockbrader H, Knapp LE. Relief of painful diabetic peripheral neuropathy with pregabalin: a randomized, placebo-controlled trial. J Pain. 2005;6(4):253–260. doi: 10.1016/j.jpain.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Pena A, Botana M, Gonzalez M, Requejo F. Expression of neurotrophins and their receptors in sciatic nerve of experimentally diabetic rats. Neuroscience letters. 1995;200(1):37–40. doi: 10.1016/0304-3940(95)12067-e. [DOI] [PubMed] [Google Scholar]
- Rosenstock J, Tuchman M, LaMoreaux L, Sharma U. Pregabalin for the treatment of painful diabetic peripheral neuropathy: a double-blind, placebo-controlled trial. Pain. 2004;110(3):628–638. doi: 10.1016/j.pain.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Rowbotham MC, Goli V, Kunz NR, Lei D. Venlafaxine extended release in the treatment of painful diabetic neuropathy: a double-blind, placebo-controlled study. Pain. 2004;110(3):697–706. doi: 10.1016/j.pain.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Rull JA, Quibrera R, Gonzalez-Millan H, Lozano Castaneda O. Symptomatic treatment of peripheral diabetic neuropathy with carbamazepine (Tegretol): double blind crossover trial. Diabetologia. 1969;5(4):215–218. doi: 10.1007/BF01212087. [DOI] [PubMed] [Google Scholar]
- Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiology of disease. 1999;6(5):347–363. doi: 10.1006/nbdi.1999.0254. [DOI] [PubMed] [Google Scholar]
- Russo VC, Gluckman PD, Feldman EL, Werther GA. The insulin-like growth factor system and its pleiotropic functions in brain. Endocrine reviews. 2005;26(7):916–943. doi: 10.1210/er.2004-0024. [DOI] [PubMed] [Google Scholar]
- Safarinejad MR. Oral sildenafil in the treatment of erectile dysfunction in diabetic men: a randomized double-blind and placebo-controlled study. Journal of diabetes and its complications. 2004;18(4):205–210. doi: 10.1016/S1056-8727(03)00056-4. [DOI] [PubMed] [Google Scholar]
- Sagara M, Satoh J, Wada R, Yagihashi S, Takahashi K, Fukuzawa M, Muto G, Muto Y, Toyota T. Inhibition of development of peripheral neuropathy in streptozotocin-induced diabetic rats with N-acetylcysteine. Diabetologia. 1996;39(3):263–269. doi: 10.1007/BF00418340. [DOI] [PubMed] [Google Scholar]
- Sang CN, Booher S, Gilron I, Parada S, Max MB. Dextromethorphan and memantine in painful diabetic neuropathy and postherpetic neuralgia: efficacy and dose-response trials. Anesthesiology. 2002;96(5):1053–1061. doi: 10.1097/00000542-200205000-00005. [DOI] [PubMed] [Google Scholar]
- Sango K, Verdes JM, Hikawa N, Horie H, Tanaka S, Inoue S, Sotelo JR, Takenaka T. Nerve growth factor (NGF) restores depletions of calcitonin gene-related peptide and substance P in sensory neurons from diabetic mice in vitro. Journal of the neurological sciences. 1994;126(1):1–5. doi: 10.1016/0022-510x(94)90087-6. [DOI] [PubMed] [Google Scholar]
- Saudek CD, Werns S, Reidenberg MM. Phenytoin in the treatment of diabetic symmetrical polyneuropathy. Clinical pharmacology and therapeutics. 1977;22(2):196–199. doi: 10.1002/cpt1977222196. [DOI] [PubMed] [Google Scholar]
- Sawicki PT, Dahne R, Bender R, Berger M. Prolonged QT interval as a predictor of mortality in diabetic nephropathy. Diabetologia. 1996;39(1):77–81. doi: 10.1007/BF00400416. [DOI] [PubMed] [Google Scholar]
- Sayeski PP, Kudlow JE. Glucose metabolism to glucosamine is necessary for glucose stimulation of transforming growth factor-alpha gene transcription. The Journal of biological chemistry. 1996;271(25):15237–15243. doi: 10.1074/jbc.271.25.15237. [DOI] [PubMed] [Google Scholar]
- Sayyed SG, Kumar A, Sharma SS. Effects of U83836E on nerve functions, hyperalgesia and oxidative stress in experimental diabetic neuropathy. Life sciences. 2006;79(8):777–783. doi: 10.1016/j.lfs.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Schmid H, Cohen CD, Henger A, Irrgang S, Schlondorff D, Kretzler M. Validation of endogenous controls for gene expression analysis in microdissected human renal biopsies. Kidney Int. 2003a;64(1):356–360. doi: 10.1046/j.1523-1755.2003.00074.x. [DOI] [PubMed] [Google Scholar]
- Schmid H, Henger A, Cohen CD, Frach K, Grone HJ, Schlondorff D, Kretzler M. Gene expression profiles of podocyte-associated molecules as diagnostic markers in acquired proteinuric diseases. J Am Soc Nephrol. 2003b;14(11):2958–2966. doi: 10.1097/01.asn.0000090745.85482.06. [DOI] [PubMed] [Google Scholar]
- Schmid H, Henger A, Kretzler M. Molecular approaches to chronic kidney disease. Current opinion in nephrology and hypertension. 2006;15(2):123–129. doi: 10.1097/01.mnh.0000214770.11609.fb. [DOI] [PubMed] [Google Scholar]
- Schmidt RE, Dorsey DA, Beaudet LN, Peterson RG. Analysis of the Zucker Diabetic Fatty (ZDF) type 2 diabetic rat model suggests a neurotrophic role for insulin/IGF-I in diabetic autonomic neuropathy. The American journal of pathology. 2003;163(1):21–28. doi: 10.1016/S0002-9440(10)63626-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt RE, Dorsey DA, Beaudet LN, Plurad SB, Parvin CA, Miller MS. Insulin-like growth factor I reverses experimental diabetic autonomic neuropathy. The American journal of pathology. 1999;155(5):1651–1660. doi: 10.1016/S0002-9440(10)65480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt RE, Dorsey DA, Beaudet LN, Plurad SB, Parvin CA, Ohara S. Effect of IGF-I and neurotrophin-3 on gracile neuroaxonal dystrophy in diabetic and aging rats. Brain research. 2000;876(1–2):88–94. doi: 10.1016/s0006-8993(00)02602-0. [DOI] [PubMed] [Google Scholar]
- Schmidt RE, Modert CW, Yip HK, Johnson EM., Jr Retrograde axonal transport of intravenously administered 125I-nerve growth factor in rats with streptozotocin-induced diabetes. Diabetes. 1983;32(7):654–663. doi: 10.2337/diab.32.7.654. [DOI] [PubMed] [Google Scholar]
- Schmidt RE, Plurad SB, Saffitz JE, Grabau GG, Yip HK. Retrograde axonal transport of 125I-nerve growth factor in rat ileal mesenteric nerves. Effect of streptozocin diabetes. Diabetes. 1985;34(12):1230–1240. doi: 10.2337/diab.34.12.1230. [DOI] [PubMed] [Google Scholar]
- Schwartz JP, Pearson J, Johnson EM. Effect of exposure to anti-NGF on sensory neurons of adult rats and guinea pigs. Brain research. 1982;244(2):378–381. doi: 10.1016/0006-8993(82)90102-0. [DOI] [PubMed] [Google Scholar]
- Sharma S, Kulkarni SK, Chopra K. Resveratrol, a polyphenolic phytoalexin attenuates thermal hyperalgesia and cold allodynia in STZ-induced diabetic rats. Indian journal of experimental biology. 2006;44(7):566–569. [PubMed] [Google Scholar]
- Sharma S, Kulkarni SK, Chopra K. Effect of resveratrol, a polyphenolic phytoalexin, on thermal hyperalgesia in a mouse model of diabetic neuropathic pain. Fundamental & clinical pharmacology. 2007;21(1):89–94. doi: 10.1111/j.1472-8206.2006.00455.x. [DOI] [PubMed] [Google Scholar]
- Sima AA, Bril V, Nathaniel V, McEwen TA, Brown MB, Lattimer SA, Greene DA. Regeneration and repair of myelinated fibers in sural-nerve biopsy specimens from patients with diabetic neuropathy treated with sorbinil. The New England journal of medicine. 1988;319(9):548–555. doi: 10.1056/NEJM198809013190905. [DOI] [PubMed] [Google Scholar]
- Sima AA, Dunlap JA, Davidson EP, Wiese TJ, Lightle RL, Greene DA, Yorek MA. Supplemental myo-inositol prevents L-fucose-induced diabetic neuropathy. Diabetes. 1997;46(2):301–306. doi: 10.2337/diab.46.2.301. [DOI] [PubMed] [Google Scholar]
- Sima AA, Zhang W, Li ZG, Murakawa Y, Pierson CR. Molecular alterations underlie nodal and paranodal degeneration in type 1 diabetic neuropathy and are prevented by C-peptide. Diabetes. 2004;53(6):1556–1563. doi: 10.2337/diabetes.53.6.1556. [DOI] [PubMed] [Google Scholar]
- Sima AA, Zhang W, Sugimoto K, Henry D, Li Z, Wahren J, Grunberger G. C-peptide prevents and improves chronic Type I diabetic polyneuropathy in the BB/Wor rat. Diabetologia. 2001;44(7):889–897. doi: 10.1007/s001250100570. [DOI] [PubMed] [Google Scholar]
- Simmons Z, Feldman EL, Williams R, Herman W, Kinmonth AL, Wareham NJ. The Evidence Base for Diabetes Care. West Sussex, England: John Wiley & Sons; 2002. Treatment of diabetic neuropathy; pp. 555–576. [Google Scholar]
- Sindrup SH, Andersen G, Madsen C, Smith T, Brosen K, Jensen TS. Tramadol relieves pain and allodynia in polyneuropathy: a randomised, double-blind, controlled trial. Pain. 1999a;83(1):85–90. doi: 10.1016/s0304-3959(99)00079-2. [DOI] [PubMed] [Google Scholar]
- Sindrup SH, Bjerre U, Dejgaard A, Brosen K, Aaes-Jorgensen T, Gram LF. The selective serotonin reuptake inhibitor citalopram relieves the symptoms of diabetic neuropathy. Clinical pharmacology and therapeutics. 1992;52(5):547–552. doi: 10.1038/clpt.1992.183. [DOI] [PubMed] [Google Scholar]
- Sindrup SH, Ejlertsen B, Froland A, Sindrup EH, Brosen K, Gram LF. Imipramine treatment in diabetic neuropathy: relief of subjective symptoms without changes in peripheral and autonomic nerve function. European journal of clinical pharmacology. 1989;37(2):151–153. doi: 10.1007/BF00558223. [DOI] [PubMed] [Google Scholar]
- Sindrup SH, Gram LF, Brosen K, Eshoj O, Mogensen EF. The selective serotonin reuptake inhibitor paroxetine is effective in the treatment of diabetic neuropathy symptoms. Pain. 1990a;42(2):135–144. doi: 10.1016/0304-3959(90)91157-E. [DOI] [PubMed] [Google Scholar]
- Sindrup SH, Gram LF, Skjold T, Froland A, Beck-Nielsen H. Concentration-response relationship in imipramine treatment of diabetic neuropathy symptoms. Clinical pharmacology and therapeutics. 1990b;47(4):509–515. doi: 10.1038/clpt.1990.65. [DOI] [PubMed] [Google Scholar]
- Sindrup SH, Gram LF, Skjold T, Grodum E, Brosen K, Beck-Nielsen H. Clomipramine vs desipramine vs placebo in the treatment of diabetic neuropathy symptoms. A double-blind cross-over study. British journal of clinical pharmacology. 1990c;30(5):683–691. doi: 10.1111/j.1365-2125.1990.tb03836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999b;83(3):389–400. doi: 10.1016/S0304-3959(99)00154-2. [DOI] [PubMed] [Google Scholar]
- Singh N, Armstrong DG, Lipsky BA. Preventing foot ulcers in patients with diabetes. Jama. 2005;293(2):217–228. doi: 10.1001/jama.293.2.217. [DOI] [PubMed] [Google Scholar]
- Singhal A, Cheng C, Sun H, Zochodne DW. Near nerve local insulin prevents conduction slowing in experimental diabetes. Brain research. 1997;763(2):209–214. doi: 10.1016/s0006-8993(97)00412-5. [DOI] [PubMed] [Google Scholar]
- Singleton JR, Smith AG, Russell J, Feldman EL. Polyneuropathy with Impaired Glucose Tolerance: Implications for Diagnosis and Therapy. Current treatment options in neurology. 2005;7(1):33–42. doi: 10.1007/s11940-005-0004-4. [DOI] [PubMed] [Google Scholar]
- Singleton JR, Smith AG, Russell JW, Feldman EL. Microvascular complications of impaired glucose tolerance. Diabetes. 2003;52(12):2867–2873. doi: 10.2337/diabetes.52.12.2867. [DOI] [PubMed] [Google Scholar]
- Soulis T, Cooper ME, Vranes D, Bucala R, Jerums G. Effects of aminoguanidine in preventing experimental diabetic nephropathy are related to the duration of treatment. Kidney Int. 1996;50(2):627–634. doi: 10.1038/ki.1996.358. [DOI] [PubMed] [Google Scholar]
- Southan GJ, Szabo C. Poly(ADP-ribose) polymerase inhibitors. Current medicinal chemistry. 2003;10(4):321–340. doi: 10.2174/0929867033368376. [DOI] [PubMed] [Google Scholar]
- Stevens MJ, Li F, Drel VR, Abatan OI, Kim H, Burnett D, Larkin D, Obrosova IG. Nicotinamide reverses neurological and neurovascular deficits in streptozotocin diabetic rats. The Journal of pharmacology and experimental therapeutics. 2007;320(1):458–464. doi: 10.1124/jpet.106.109702. [DOI] [PubMed] [Google Scholar]
- Stracke H, Lindemann A, Federlin K. A benfotiamine-vitamin B combination in treatment of diabetic polyneuropathy. Exp Clin Endocrinol Diabetes. 1996;104(4):311–316. doi: 10.1055/s-0029-1211460. [DOI] [PubMed] [Google Scholar]
- Stracke H, Meyer UE, Schumacher HE, Federlin K. Mexiletine in the treatment of diabetic neuropathy. Diabetes care. 1992;15(11):1550–1555. doi: 10.2337/diacare.15.11.1550. [DOI] [PubMed] [Google Scholar]
- Stribling D, Mirrlees DJ, Harrison HE, Earl DC. Properties of ICI 128,436, a novel aldose reductase inhibitor, and its effects on diabetic complications in the rat. Metabolism: clinical and experimental. 1985;34(4):336–344. doi: 10.1016/0026-0495(85)90223-9. [DOI] [PubMed] [Google Scholar]
- Strokov IA, Manukhina EB, Bakhtina LY, Malyshev IY, Zoloev GK, Kazikhanova SI, Ametov AS. The function of endogenous protective systems in patients with insulin-dependent diabetes mellitus and polyneuropathy: effect of antioxidant therapy. Bulletin of experimental biology and medicine. 2000;130(10):986–990. [PubMed] [Google Scholar]
- Sugimoto K, Murakawa Y, Sima AA. Expression and localization of insulin receptor in rat dorsal root ganglion and spinal cord. J Peripher Nerv Syst. 2002;7(1):44–53. doi: 10.1046/j.1529-8027.2002.02005.x. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Murakawa Y, Zhang W, Xu G, Sima AA. Insulin receptor in rat peripheral nerve: its localization and alternatively spliced isoforms. Diabetes/metabolism research and reviews. 2000;16(5):354–363. doi: 10.1002/1520-7560(200009/10)16:5<354::aid-dmrr149>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Sullivan KA, Feldman EL. New developments in diabetic neuropathy. Current opinion in neurology. 2005;18(5):586–590. doi: 10.1097/01.wco.0000178825.56414.52. [DOI] [PubMed] [Google Scholar]
- Sullivan KA, Hayes JM, Wiggin TD, Backus C, Su Oh S, Lentz SI, Brosius F, 3rd, Feldman EL. Mouse models of diabetic neuropathy. Neurobiology of disease. 2007;28(3):276–285. doi: 10.1016/j.nbd.2007.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KA, Lentz SI, Roberts JL, Jr, Feldman EL. Criteria for creating and assessing mouse models of diabetic neuropathy. Current drug targets. 2008;9(1):3–13. doi: 10.2174/138945008783431763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, Zhai Q. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell metabolism. 2007;6(4):307–319. doi: 10.1016/j.cmet.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Tamasi V, Jeffries JM, Arteel GE, Falkner KC. Ebselen augments its peroxidase activity by inducing nrf-2-dependent transcription. Archives of biochemistry and biophysics. 2004;431(2):161–168. doi: 10.1016/j.abb.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A, Pischetsrieder M, Stern D, Schmidt AM, D’Agati VD. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol. 2000;11(9):1656–1666. doi: 10.1681/ASN.V1191656. [DOI] [PubMed] [Google Scholar]
- Tankova T, Koev D, Dakovska L. Alpha-lipoic acid in the treatment of autonomic diabetic neuropathy (controlled, randomized, open-label study) Romanian journal of internal medicine = Revue roumaine de medecine interne. 2004;42(2):457–464. [PubMed] [Google Scholar]
- Tesch GH. Role of macrophages in complications of type 2 diabetes. Clinical and experimental pharmacology & physiology. 2007;34(10):1016–1019. doi: 10.1111/j.1440-1681.2007.04729.x. [DOI] [PubMed] [Google Scholar]
- Tesfaye S, Chaturvedi N, Eaton SE, Ward JD, Manes C, Ionescu-Tirgoviste C, Witte DR, Fuller JH. Vascular risk factors and diabetic neuropathy. The New England journal of medicine. 2005;352(4):341–350. doi: 10.1056/NEJMoa032782. [DOI] [PubMed] [Google Scholar]
- Tesfaye S, Stevens LK, Stephenson JM, Fuller JH, Plater M, Ionescu-Tirgoviste C, Nuber A, Pozza G, Ward JD. Prevalence of diabetic peripheral neuropathy and its relation to glycaemic control and potential risk factors: the EURODIAB IDDM Complications Study. Diabetologia. 1996;39(11):1377–1384. doi: 10.1007/s001250050586. [DOI] [PubMed] [Google Scholar]
- Tesfaye S, Tandan R, Bastyr EJ, 3rd, Kles KA, Skljarevski V, Price KL. Factors that impact symptomatic diabetic peripheral neuropathy in placebo-administered patients from two 1-year clinical trials. Diabetes care. 2007;30(10):2626–2632. doi: 10.2337/dc07-0608. [DOI] [PubMed] [Google Scholar]
- Thamotharampillai K, Chan AK, Bennetts B, Craig ME, Cusumano J, Silink M, Oates PJ, Donaghue KC. Decline in neurophysiological function after 7 years in an adolescent diabetic cohort and the role of aldose reductase gene polymorphisms. Diabetes care. 2006;29(9):2053–2057. doi: 10.2337/dc06-0678. [DOI] [PubMed] [Google Scholar]
- The Capsaicin Study G. Effect of treatment with capsaicin on daily activities of patients with painful diabetic neuropathy. Diabetes care. 1992;15:159–165. doi: 10.2337/diacare.15.2.159. [DOI] [PubMed] [Google Scholar]
- Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Research. 2002;62:5196–5203. [PubMed] [Google Scholar]
- Thomas PK. Diabetic peripheral neuropathies: their cost to patient and society and the value of knowledge of risk factors for development of interventions. European neurology. 1999;41(Suppl 1):35–43. doi: 10.1159/000052078. [DOI] [PubMed] [Google Scholar]
- Thornalley PJ. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Archives of biochemistry and biophysics. 2003;419(1):31–40. doi: 10.1016/j.abb.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Thornalley PJ. The potential role of thiamine (vitamin B(1)) in diabetic complications. Current diabetes reviews. 2005;1(3):287–298. doi: 10.2174/157339905774574383. [DOI] [PubMed] [Google Scholar]
- Toth C, Brussee V, Zochodne DW. Remote neurotrophic support of epidermal nerve fibres in experimental diabetes. Diabetologia. 2006;49(5):1081–1088. doi: 10.1007/s00125-006-0169-8. [DOI] [PubMed] [Google Scholar]
- Toth C, Rong LL, Yang C, Martinez J, Song F, Ramji N, Brussee V, Liu W, Durand J, Nguyen MD, Schmidt AM, Zochodne DW. RAGE and Experimental Diabetic Neuropathy. Diabetes. 2007 doi: 10.2337/db07-0339. [DOI] [PubMed] [Google Scholar]
- Traber MG, Packer L. Vitamin E: beyond antioxidant function. The American journal of clinical nutrition. 1995;62(6 Suppl):1501S–1509S. doi: 10.1093/ajcn/62.6.1501S. [DOI] [PubMed] [Google Scholar]
- Traber MG, Sies H. Vitamin E in humans: demand and delivery. Annual review of nutrition. 1996;16:321–347. doi: 10.1146/annurev.nu.16.070196.001541. [DOI] [PubMed] [Google Scholar]
- Triantafillidis JK, Kottaras G, Sgourous S, Cheracakis P, Driva G, Konstantellou E, Parasi A, Choremi H, Samouilidou E. A-beta-lipoproteinemia: clinical and laboratory features, therapeutic manipulations, and follow-up study of three members of a Greek family. Journal of clinical gastroenterology. 1998;26(3):207–211. doi: 10.1097/00004836-199804000-00012. [DOI] [PubMed] [Google Scholar]
- Trotta D, Verrotti A, Salladini C, Chiarelli F. Diabetic neuropathy in children and adolescents. Pediatric diabetes. 2004;5(1):44–57. doi: 10.1111/j.1399-543X.2004.00041.x. [DOI] [PubMed] [Google Scholar]
- Tuttle KR, Bakris GL, Toto RD, McGill JB, Hu K, Anderson PW. The effect of ruboxistaurin on nephropathy in type 2 diabetes. Diabetes care. 2005;28(11):2686–2690. doi: 10.2337/diacare.28.11.2686. [DOI] [PubMed] [Google Scholar]
- Tutuncu NB, Batur MK, Yildirir A, Tutuncu T, Deger A, Koray Z, Erbas B, Kabakci G, Aksoyek S, Erbas T. Melatonin levels decrease in type 2 diabetic patients with cardiac autonomic neuropathy. Journal of pineal research. 2005;39(1):43–49. doi: 10.1111/j.1600-079X.2005.00213.x. [DOI] [PubMed] [Google Scholar]
- Uehara K, Yamagishi S, Otsuki S, Chin S, Yagihashi S. Effects of polyol pathway hyperactivity on protein kinase C activity, nociceptive peptide expression, and neuronal structure in dorsal root ganglia in diabetic mice. Diabetes. 2004;53(12):3239–3247. doi: 10.2337/diabetes.53.12.3239. [DOI] [PubMed] [Google Scholar]
- Unger JW, Klitzsch T, Pera S, Reiter R. Nerve growth factor (NGF) and diabetic neuropathy in the rat: morphological investigations of the sural nerve, dorsal root ganglion, and spinal cord. Experimental neurology. 1998;153(1):23–34. doi: 10.1006/exnr.1998.6856. [DOI] [PubMed] [Google Scholar]
- van Houtum WH, Rauwerda JA, Ruwaard D, Schaper NC, Bakker K. Reduction in diabetes-related lower-extremity amputations in The Netherlands: 1991–2000. Diabetes care. 2004;27(5):1042–1046. doi: 10.2337/diacare.27.5.1042. [DOI] [PubMed] [Google Scholar]
- Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17(24):3145–3156. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- Vergara C, Ramirez B. CNTF, a pleiotropic cytokine: emphasis on its myotrophic role. Brain Res Brain Res Rev. 2004;47(1–3):161–173. doi: 10.1016/j.brainresrev.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Verrotti A, Chiarelli F, Blasetti A, Morgese G. Autonomic neuropathy in diabetic children. Journal of paediatrics and child health. 1995;31(6):545–548. doi: 10.1111/j.1440-1754.1995.tb00881.x. [DOI] [PubMed] [Google Scholar]
- Veves A, King GL. Can VEGF reverse diabetic neuropathy in human subjects? The Journal of clinical investigation. 2001;107(10):1215–1218. doi: 10.1172/JCI13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vileikyte L, Peyrot M, Bundy C, Rubin RR, Leventhal H, Mora P, Shaw JE, Baker P, Boulton AJ. The development and validation of a neuropathy- and foot ulcer-specific quality of life instrument. Diabetes care. 2003;26(9):2549–2555. doi: 10.2337/diacare.26.9.2549. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Edwards JL, Sadidi M, Feldman EL. The antioxidant response as a drug target in diabetic neuropathy. Current drug targets. 2008;9(1):94–100. doi: 10.2174/138945008783431754. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Feldman EL. New insights into the mechanisms of diabetic neuropathy. Reviews in endocrine & metabolic disorders. 2004a;5(3):227–236. doi: 10.1023/B:REMD.0000032411.11422.e0. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Perrone L, Sullivan KA, Backus C, Sastry AM, Lastoskie C, Feldman EL. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology. 2007;148(2):548–558. doi: 10.1210/en.2006-0073. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocrine reviews. 2004b;25(4):612–628. doi: 10.1210/er.2003-0019. [DOI] [PubMed] [Google Scholar]
- Vinik A, Mehrabyan A, Colen L, Boulton A. Focal entrapment neuropathies in diabetes. Diabetes care. 2004a;27(7):1783–1788. doi: 10.2337/diacare.27.7.1783. [DOI] [PubMed] [Google Scholar]
- Vinik AI, Bril V, Kempler P, Litchy WJ, Tesfaye S, Price KL, Bastyr EJ., 3rd Treatment of symptomatic diabetic peripheral neuropathy with the protein kinase C beta-inhibitor ruboxistaurin mesylate during a 1-year, randomized, placebo-controlled, double-blind clinical trial. Clinical therapeutics. 2005;27(8):1164–1180. doi: 10.1016/j.clinthera.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Vinik AI, Maser RE, Mitchell BD, Freeman R. Diabetic autonomic neuropathy. Diabetes care. 2003;26(5):1553–1579. doi: 10.2337/diacare.26.5.1553. [DOI] [PubMed] [Google Scholar]
- Vinik AI, Mehrabyan A. Diabetic neuropathies. The Medical clinics of North America. 2004b;88(4):947–999. xi. doi: 10.1016/j.mcna.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Vinik AI, Tuchman M, Safirstein B, Corder C, Kirby L, Wilks K, Quessy S, Blum D, Grainger J, White J, Silver M. Lamotrigine for treatment of pain associated with diabetic neuropathy: results of two randomized, double-blind, placebo-controlled studies. Pain. 2007;128(1–2):169–179. doi: 10.1016/j.pain.2006.09.040. [DOI] [PubMed] [Google Scholar]
- Wada R, Nishizawa Y, Yagihashi N, Takeuchi M, Ishikawa Y, Yasumura K, Nakano M, Yagihashi S. Effects of OPB-9195, anti-glycation agent, on experimental diabetic neuropathy. European journal of clinical investigation. 2001;31(6):513–520. doi: 10.1046/j.1365-2362.2001.00826.x. [DOI] [PubMed] [Google Scholar]
- Wada R, Yagihashi S. Role of advanced glycation end products and their receptors in development of diabetic neuropathy. Annals of the New York Academy of Sciences. 2005;1043:598–604. doi: 10.1196/annals.1338.067. [DOI] [PubMed] [Google Scholar]
- Wahren J, Shafqat J, Johansson J, Chibalin A, Ekberg K, Jornvall H. Molecular and cellular effects of C-peptide--new perspectives on an old peptide. Experimental diabesity research. 2004;5(1):15–23. doi: 10.1080/15438600490424479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Schmeichel AM, Iida H, Schmelzer JD, Low PA. Enhanced inflammatory response via activation of NF-kappaB in acute experimental diabetic neuropathy subjected to ischemia-reperfusion injury. Journal of the neurological sciences. 2006;247(1):47–52. doi: 10.1016/j.jns.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Watson CP, Moulin D, Watt-Watson J, Gordon A, Eisenhoffer J. Controlled-release oxycodone relieves neuropathic pain: a randomized controlled trial in painful diabetic neuropathy. Pain. 2003;105(1–2):71–78. doi: 10.1016/s0304-3959(03)00160-x. [DOI] [PubMed] [Google Scholar]
- Wear-Maggitti K, Lee J, Conejero A, Schmidt AM, Grant R, Breitbart A. Use of topical sRAGE in diabetic wounds increases neovascularization and granulation tissue formation. Annals of plastic surgery. 2004;52(5):519–521. doi: 10.1097/01.sap.0000122857.49274.8c. discussion 522. [DOI] [PubMed] [Google Scholar]
- Wernicke JF, Pritchett YL, D’Souza DN, Waninger A, Tran P, Iyengar S, Raskin J. A randomized controlled trial of duloxetine in diabetic peripheral neuropathic pain. Neurology. 2006;67:1411–1420. doi: 10.1212/01.wnl.0000240225.04000.1a. [DOI] [PubMed] [Google Scholar]
- Wiffen P, Collins S, McQuay H, Carroll D, Jadad A, Moore A. Anticonvulsant drugs for acute and chronic pain. Cochrane database of systematic reviews (Online) 2005;(3):CD001133. doi: 10.1002/14651858.CD001133.pub2. [DOI] [PubMed] [Google Scholar]
- Will JC, Ford ES, Bowman BA. Serum vitamin C concentrations and diabetes: findings from the Third National Health and Nutrition Examination Survey, 1988–1994. The American journal of clinical nutrition. 1999;70(1):49–52. doi: 10.1093/ajcn/70.1.49. [DOI] [PubMed] [Google Scholar]
- Williams B, Gallacher B, Patel H, Orme C. Glucose-induced protein kinase C activation regulates vascular permeability factor mRNA expression and peptide production by human vascular smooth muscle cells in vitro. Diabetes. 1997;46(9):1497–1503. doi: 10.2337/diab.46.9.1497. [DOI] [PubMed] [Google Scholar]
- Wilson DM, Block J. Real-time continuous glucose monitor use and patient selection: what have we learned and where are we going? Diabetes technology & therapeutics. 2005;7(5):788–791. doi: 10.1089/dia.2005.7.788. [DOI] [PubMed] [Google Scholar]
- Windebank AJ, Feldman EL, Aminoff MJ. Neurology and General Medicine. Vol. 3. Churchill Livingstone; 2001. Diabetes and the nervous system; pp. 341–364. [Google Scholar]
- Winkler G, Pal B, Nagybeganyi E, Ory I, Porochnavec M, Kempler P. Effectiveness of different benfotiamine dosage regimens in the treatment of painful diabetic neuropathy. Arzneimittel-Forschung. 1999;49(3):220–224. doi: 10.1055/s-0031-1300405. [DOI] [PubMed] [Google Scholar]
- Wozniak K, Arabski M, Malecka-Panas E, Drzewoski J, Blasiak J. DNA damage in human colonic mucosa cells induced by bleomycin and the protective action of vitamin E. Cellular & molecular biology letters. 2004;9(1):31–45. [PubMed] [Google Scholar]
- Wuarin L, Guertin DM, Ishii DN. Early reduction in insulin-like growth factor gene expression in diabetic nerve. Experimental neurology. 1994;130(1):106–114. doi: 10.1006/exnr.1994.1189. [DOI] [PubMed] [Google Scholar]
- Xia P, Kramer RM, King GL. Identification of the mechanism for the inhibition of Na+, K(+)-adenosine triphosphatase by hyperglycemia involving activation of protein kinase C and cytosolic phospholipase A2. The Journal of clinical investigation. 1995;96(2):733–740. doi: 10.1172/JCI118117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagihashi S, Kamijo M, Ido Y, Mirrlees DJ. Effects of long-term aldose reductase inhibition on development of experimental diabetic neuropathy. Ultrastructural and morphometric studies of sural nerve in streptozocin-induced diabetic rats. Diabetes. 1990;39(6):690–696. doi: 10.2337/diab.39.6.690. [DOI] [PubMed] [Google Scholar]
- Yamagishi S, Ogasawara S, Mizukami H, Yajima N, Wada R, Sugawara A, Yagihashi S. Correction of protein kinase C activity and macrophage migration in peripheral nerve by pioglitazone, peroxisome proliferator activated-gamma-ligand, in insulin-deficient diabetic rats. Journal of neurochemistry. 2008;104(2):491–499. doi: 10.1111/j.1471-4159.2007.05050.x. [DOI] [PubMed] [Google Scholar]
- Yamagishi S, Uehara K, Otsuki S, Yagihashi S. Differential influence of increased polyol pathway on protein kinase C expressions between endoneurial and epineurial tissues in diabetic mice. Journal of neurochemistry. 2003;87(2):497–507. doi: 10.1046/j.1471-4159.2003.02011.x. [DOI] [PubMed] [Google Scholar]
- Yang S, Litchfield JE, Baynes JW. AGE-breakers cleave model compounds, but do not break Maillard crosslinks in skin and tail collagen from diabetic rats. Archives of biochemistry and biophysics. 2003;412(1):42–46. doi: 10.1016/s0003-9861(03)00015-8. [DOI] [PubMed] [Google Scholar]
- Yao D, Taguchi T, Matsumura T, Pestell R, Edelstein D, Giardino I, Suske G, Rabbani N, Thornalley PJ, Sarthy VP, Hammes HP, Brownlee M. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. The Journal of biological chemistry. 2007;282(42):31038–31045. doi: 10.1074/jbc.M704703200. [DOI] [PubMed] [Google Scholar]
- Young MJ, Boulton AJ, MacLeod AF, Williams DR, Sonksen PH. A multicentre study of the prevalence of diabetic peripheral neuropathy in the United Kingdom hospital clinic population. Diabetologia. 1993;36(2):150–154. doi: 10.1007/BF00400697. [DOI] [PubMed] [Google Scholar]
- Yuen KC, Baker NR, Rayman G. Treatment of chronic painful diabetic neuropathy with isosorbide dinitrate spray: a double-blind placebo-controlled cross-over study. Diabetes care. 2002;25(10):1699–1703. doi: 10.2337/diacare.25.10.1699. [DOI] [PubMed] [Google Scholar]
- Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, Wierzbicki M, Verbeuren TJ, Cohen RA. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;55(8):2180–2191. doi: 10.2337/db05-1188. [DOI] [PubMed] [Google Scholar]
- Zhang W, Kamiya H, Ekberg K, Wahren J, Sima AA. C-peptide improves neuropathy in type 1 diabetic BB/Wor-rats. Diabetes/metabolism research and reviews. 2007;23(1):63–70. doi: 10.1002/dmrr.672. [DOI] [PubMed] [Google Scholar]
- Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53(11):2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]
- Zhuang HX, Wuarin L, Fei ZJ, Ishii DN. Insulin-like growth factor (IGF) gene expression is reduced in neural tissues and liver from rats with non-insulin-dependent diabetes mellitus, and IGF treatment ameliorates diabetic neuropathy. The Journal of pharmacology and experimental therapeutics. 1997;283(1):366–374. [PubMed] [Google Scholar]
- Ziegler D. Treatment of diabetic polyneuropathy: Update 2006. Annals of the New York Academy of Sciences. 2006;1084:250–266. doi: 10.1196/annals.1372.008. [DOI] [PubMed] [Google Scholar]
- Ziegler D, Gries FA, Muhlen H, Rathmann W, Spuler M, Lessmann F. Prevalence and clinical correlates of cardiovascular autonomic and peripheral diabetic neuropathy in patients attending diabetes centers. The Diacan Multicenter Study Group. Diabete & metabolisme. 1993;19(1 Pt 2):143–151. [PubMed] [Google Scholar]
- Ziegler D, Hanefeld M, Ruhnau KJ, Hasche H, Lobisch M, Schutte K, Kerum G, Malessa R. Treatment of symptomatic diabetic polyneuropathy with the antioxidant alpha-lipoic acid: a 7-month multicenter randomized controlled trial (ALADIN III Study). ALADIN III Study Group. Alpha-Lipoic Acid in Diabetic Neuropathy. Diabetes care. 1999a;22(8):1296–1301. doi: 10.2337/diacare.22.8.1296. [DOI] [PubMed] [Google Scholar]
- Ziegler D, Reljanovic M, Mehnert H, Gries FA. Alpha-lipoic acid in the treatment of diabetic polyneuropathy in Germany: current evidence from clinical trials. Exp Clin Endocrinol Diabetes. 1999b;107(7):421–430. doi: 10.1055/s-0029-1212132. [DOI] [PubMed] [Google Scholar]
- Ziegler D, Schatz H, Conrad F, Gries FA, Ulrich H, Reichel G. Effects of treatment with the antioxidant alpha-lipoic acid on cardiac autonomic neuropathy in NIDDM patients. A 4-month randomized controlled multicenter trial (DEKAN Study). Deutsche Kardiale Autonome Neuropathie. Diabetes care. 1997;20(3):369–373. doi: 10.2337/diacare.20.3.369. [DOI] [PubMed] [Google Scholar]
- Zieman SJ, Melenovsky V, Clattenburg L, Corretti MC, Capriotti A, Gerstenblith G, Kass DA. Advanced glycation endproduct crosslink breaker (alagebrium) improves endothelial function in patients with isolated systolic hypertension. Journal of hypertension. 2007;25(3):577–583. doi: 10.1097/HJH.0b013e328013e7dd. [DOI] [PubMed] [Google Scholar]
- Zochodne DW, Levy D. Nitric oxide in damage, disease and repair of the peripheral nervous system. Cellular and molecular biology (Noisy-le-Grand, France) 2005;51(3):255–267. [PubMed] [Google Scholar]