
Assembly of large genomes using second-generation sequencing
- Center for Bioinformatics and Computational Biology, University of Maryland, College Park, Maryland 20742, USA
Abstract
Second-generation sequencing technology can now be used to sequence an entire human genome in a matter of days and at low cost. Sequence read lengths, initially very short, have rapidly increased since the technology first appeared, and we now are seeing a growing number of efforts to sequence large genomes de novo from these short reads. In this Perspective, we describe the issues associated with short-read assembly, the different types of data produced by second-gen sequencers, and the latest assembly algorithms designed for these data. We also review the genomes that have been assembled recently from short reads and make recommendations for sequencing strategies that will yield a high-quality assembly.
As genome sequencing technology has evolved, methods for assembling genomes have changed with it. Genome sequencers have never been able to “read” more than a relatively short stretch of DNA at once, with read lengths gradually increasing over time. Reconstructing a complete genome from a set of reads requires an assembly program, and a variety of genome assemblers have been used for this task. In 1995, when the first bacterial genome was published (Haemophilus influenzae), read lengths were ∼460 base pairs (bp), and that whole-genome shotgun (WGS) sequencing project generated 24,304 reads (Fleischmann et al. 1995). The human genome project required ∼30 million reads, with lengths up to 800 bp, using Sanger sequencing technology and automated capillary sequencers (International Human Genome Sequencing Consortium 2001; Venter et al. 2001). This corresponded to 24 billion bases (Gb), or approximately eightfold coverage of the 3-Gb human genome. Redundant coverage, in which on average every nucleotide is sequenced many times over, is required to produce a high-quality assembly. Another benefit of redundancy is greatly increased accuracy compared with a single read: Where a single read might have an error rate of 1%, eightfold coverage has an error rate as low as 10−16 when eight high-quality reads agree with one another. High coverage is also necessary to sequence polymorphic alleles within diploid or polyploid genomes.
Current second-generation sequencing (SGS) technologies produce read lengths ranging from 35 to 400 bp, at far greater speed and much lower cost than Sanger sequencing. However, as reads get shorter, coverage needs to increase to compensate for the decreased connectivity and produce a comparable assembly. Certain problems cannot be overcome by deeper coverage: If a repetitive sequence is longer than a read, then coverage alone will never compensate, and all copies of that sequence will produce gaps in the assembly. These gaps can be spanned by paired reads—consisting of two reads generated from a single fragment of DNA and separated by a known distance—as long as the pair separation distance is longer than the repeat. Paired-end sequencing is available from most of the SGS machines, although it is not yet as flexible or as reliable as paired-end sequencing using traditional methods.
After the successful assembly of the human (International Human Genome Sequencing Consortium 2001; Venter et al. 2001) and mouse (Waterston et al. 2002) genomes by whole-genome shotgun sequencing, most large-scale genome projects quickly moved to adopt the WGS approach, which has subsequently been used for dozens of eukaryotic genomes. Today, thanks to changes in sequencing technology, a major question confronting genome projects is, can we sequence a large genome (>100 Mbp) using short reads? If so, what are the limitations on read length, coverage, and error rates? How much paired-end sequencing is necessary? And what will the assembly look like? In this perspective we take a look at each of these questions and describe the solutions available today. Although we provide some answers, we have no doubt that the solutions will change rapidly over the next few years, as both the sequencing methods and the computational solutions improve.
Overview of SGS technologies
The two leading sequencing technologies today produce reads with decidedly different characteristics. The pyrosequencing approach, embodied in the 454 Life Sciences sequencer from Roche, produces read lengths approaching 400 bp, and in a single 1-d run generates several hundred million nucleotides. This technology sequences DNA by sequentially flowing bases in a predetermined order across templates that are captured on microscopic beads contained in tiny wells. A single cycle will incorporate multiple bases whenever the template sequence has a homopolymer run. Base calling is done by measuring the fluorescence intensity at each well, with greater intensity corresponding to multiple bases. Read lengths and error rates have steadily improved since this method was introduced in 2005 (Margulies et al. 2005), and 800-bp reads are expected in the near future. At that point, pyrosequencing read lengths will match those of Sanger sequencing.
The alternative approach produces shorter reads, but at much higher throughput. This approach is embodied in several different commercial sequencers, including those from Illumina, Applied Biosystems, and Helicos. The shared theme is to incorporate only one base per cycle, using specially modified bases that include both a fluorescent tag and a terminator (Schuster 2008). After reading the base with a laser, the tag and terminator are removed so that the template can be extended by one more base. These machines operate at much higher densities, produce 20–30 Gb per run, although a single run takes 5–10 d depending on the machine. Read lengths have grown over the past 2 yr from 25 to 30 bases to >100 bases today on some platforms. The overall cost per run is similar to pyrosequencing, yielding a much lower per-base cost.
All these platforms offer some form of paired-end sequencing, but thus far the reliability of paired ends is not nearly as good as it is for Sanger sequencing. In conventional Sanger sequencing, a “long” paired-end protocol starts with DNA templates ranging from 5000 to 35,000 bp. These fragments are cloned into a vector, which is then amplified in Escherichia coli prior to sequencing. The vectors are subsequently extracted and then both ends of the vector inserts are sequenced. One drawback to this traditional method is that the E. coli cloning step introduces a bias, making it difficult to capture some regions of a genome.
Paired-end protocols for SGS avoid the use of a bacterial cloning step. Instead, they generally start with DNA fragments of the desired size, and then try to sequence both ends by circularizing the DNA, using a special tag or linker to connect the ends. By sequencing fragments containing the tag, both ends of the original fragment will be captured. Although this sounds straightforward, experience to date has indicated that it is very difficult to get DNA to circularize efficiently, and problems increase as the fragments get longer (Collins and Weissman 1984). As a result, many paired-end libraries contain too little DNA, and the paired-end sequences fail to cover the genome at the required depth. Some techniques do not require circularization and are correspondingly much more reliable, but these only produce paired reads at a distance of ∼500 bp. For even longer pairs, such as those produced by fosmids or bacterial artificial chromosome (BAC) ends (30–150 kbp), no protocols currently exist. This has significant implications for genome assembly, as we discuss below.
Overview of assembly methods
Current genome sequencing technology can only sequence a tiny portion of a genome in a contiguous read. Nevertheless, just as a jigsaw puzzle can be assembled from small puzzle pieces, a complete genome sequence can be assembled from short reads. Unlike jigsaw puzzle pieces that precisely lock together, DNA sequence reads may fit together in more than one way because of repetitive sequences within the genome. Assembly methods aim to create the most complete reconstruction possible without introducing errors.
The central challenge of genome assembly is resolving repetitive sequences. The magnitude of the challenge depends on the sequencing technology, because the fraction of repetitive reads depends on the length of reads themselves. At one extreme, if the reads were just one base long, every read would be repetitive; at the other extreme, if we could simply read an entire chromosome from one end to the other, repeats would pose no problem at all. In between these extremes, the fraction of unique sequences increases as the read length increases, until eventually every sequence in the genome is unique. If DNA sequences were random (which they are not), then the expected number of occurrences of any sequence would decrease exponentially as the length of the sequence increases, and a modest increase in read length could dramatically reduce the number of repeats in the genome. However, real genomes have complicated repeat structures making some sequences nearly impossible to assembly correctly.
To illustrate the variability in repetitiveness among species, Figure 1 shows the uniqueness ratio for varying read lengths (constructed using the tallymer tool; Kurtz et al. 2008) plotted for six genomes: fruit fly (Drosophila melanogaster), grapevine (Vitis vinifera), chicken (Gallus gallus), dog (Canis familiaris), human (Homo sapiens), and the single-celled parasite Trichomonas vaginalis. The figure shows how much of each genome would be covered by k-mers (reads) that occur exactly once. Among the multicellular species, dog and chicken are the least repetitive while fly is the most repetitive. The percentage of a genome covered uniquely increases rapidly as read length increases to 50 bp and above, but the rate of increase varies due to the variable repeat lengths in different species.
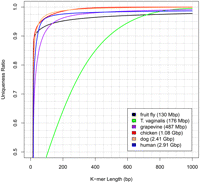
The k-mer uniqueness ratio for five well-known organisms and one single-celled human parasite. The ratio is defined here as the percentage of the genome that is covered by unique sequences of length k or longer. The horizontal axis shows the length in base pairs of the sequences. For example, ∼92.5% of the grapevine genome is contained in unique sequences of 100 bp or longer.
Early genome assemblers used a simple “greedy” algorithm, in which all pairs of reads are compared with each other, and the ones that overlap most are merged first. To allow for sequencing errors, assemblers compute these overlaps with a variant of the Smith-Waterman algorithm (Smith and Waterman 1981), which allows for a small number of differences in the overlapping sequence, typically 1%–10%. Once all overlaps are computed, the reads with the longest overlap are concatenated to form a contig (contiguous sequence). The process then repeats, each time merging the sequences with the longest overlap until all overlaps are used.
This simple merging process will accurately reconstruct the simplest genomes, but fails for repetitive sequences longer than the read length. The greedy algorithm will assemble all copies of a repeat into a single instance, because all reads with the repetitive sequence overlap equally well. The problem is that the greedy algorithm cannot tell how to connect the unique sequences on either end of a repeat, and it can easily assemble together distant portions of the genome into misassembled, “chimeric” contigs. Beginning in the 1990s, assembly of bacterial genomes required development of more sophisticated methods to handle repetitive sequences. Assembly of large eukaryotic genomes required further innovations, not only in the handling of repeats, but also in the computational requirements for memory and processing time. If these issues are not handled in a sophisticated way, then the enormous data sets comprising mammalian genome projects will simply overwhelm even the largest computers.
Large-scale shotgun assembly
Several assemblers have been developed to assemble large, repetitive genomes from long (“Sanger”) reads, including the Celera Assembler (Myers et al. 2000), ARACHNE (Batzoglou et al. 2002; Jaffe et al. 2003), and PCAP (Huang et al. 2003). More recently, the Newbler assembler (Margulies et al. 2005) was designed to handle shorter 454 Life Sciences (Roche) reads, which have a different error profile from Sanger reads. Unlike simple greedy assemblers, these algorithms assemble the reads in two or more distinct phases, with separate processing of repetitive sequences. First, they assemble reads with unambiguous overlaps, creating contigs that end on the boundaries of repeats. (Myers et al. [2000] call these “unitigs.”) Then, in a second phase, they assemble the unambiguous contigs together into larger sequences, using mate-pair constraints to resolve repeats.
As with earlier methods, these large-scale assemblers begin by computing overlaps between all pairs of reads. One technique for saving memory, used by Celera Assembler (CABOG), is to construct an overlap graph where each read is a node in the graph, and weighted edges connect overlapping reads. These assemblers also attempt to correct sequencing errors by using overlapping reads to confirm each other. These error correction methods can be very effective when coverage is deep, as it often is with newer short-read sequencing projects.
The scaffolding phase of assembly focuses on resolving repeats by linking the initial contigs into scaffolds, guided by mate-pair data. Mate pairs constrain the separation distance and the orientation of contigs containing mated reads. A scaffold is a collection of contigs linked by mate pairs, in which the gaps between contigs may represent either repeats, in which case the gap can in theory be filled with one or more copies of the repeat, or true gaps in which the original sequencing project did not capture the sequence needed to fill the gap. If the mate pair distances are long enough, they permit the assembler to link contigs across almost all repeats.
Assemblers vary in their strategies for calling a contig repetitive, but most of them rely on some combination of the length of the contig and the number of reads it contains. If a contig contains too many reads, then it is flagged as a repeat. High copy-number repeats are easy to identify, because the coverage statistics make it obvious that they are repetitive; in contrast, two-copy repeats are the most difficult to identify using statistical methods.
After flagging repeats, an assembler can build scaffolds by connecting unique contigs using mate-pair links. If the contigs in a scaffold overlap, the assembler can merge them at this point. Otherwise, the assembler will record a gap of approximately known size within the scaffold. Assemblers can also include repetitive contigs in these scaffolds, as long as the repeats are connected by mate pairs to unique contigs.
Short read assembly
In principle, assemblers created for long reads should also function for short reads. The principles of detecting overlap and building contigs are no different. In practice, initial attempts to use existing assemblers with very short reads either failed or performed very poorly, for a variety of reasons. Some of these failures were mundane: For example, assemblers impose a minimum read length, or they require a minimum amount of overlap that is too long for a short-read sequencing project. Other failures are caused by more fundamental problems.
The computation of overlaps is one of the most critical steps in any assembly algorithm. Short-read sequencing projects require that this step be redesigned to make it computationally feasible, especially since many more short reads than long reads are needed to achieve the same level of coverage. (Coverage is defined as the average number of reads that contain any nucleotide; thus, 8× coverage implies that the genome is sequenced eight times over.) As such, the number of overlaps to compute will increase, and any per-read or per-overlap overhead will be greatly magnified. This problem is exacerbated by the fact that short-read projects compensate for read length by obtaining deeper coverage, and it is not unusual to see SGS projects at 30×, 40×, or 50× coverage rather than the 8× coverage that is typical of Sanger sequencing projects.
The parameters used for computing overlaps have to be carefully tuned to accommodate shorter read lengths. Genome assemblers such as CABOG and ARACHNE do not compute the overlap between all pairs of reads, but instead use a seed-based strategy to identify reads that are likely to overlap. With this approach, short fixed length substrings of the reads, k-mers, are used as an index, and only pairs of reads that share a seed are evaluated further. The choice of seed length is critical and depends on the length of the read, the amount of sequencing error, and the size of the genome. If the seed is too long, legitimate overlaps will be missed, thereby fragmenting the assembly, but, if the seed length is too short, the computation time increases dramatically, so much that the computation may no longer be feasible. In addition to adjusting the seed length for short reads, the amount of error varies among SGS technologies, meaning that assemblers may have to be fine-tuned separately for each sequencing technology.
For these reasons and others, a new generation of genome assemblers has been developed specifically to address the challenges of assembling very short reads. These assemblers include Velvet (Zerbino and Birney 2008; Zerbino et al. 2009), ALLPATHS (Butler et al. 2008; Maccallum et al. 2009), ABySS (Simpson et al. 2009) SOAPdenovo (Li et al. 2010), and Contrail (http://contrail-bio.sf.net). Rather than using an overlap graph, all of these assemblers use a de Bruijn graph algorithm, first described for the EULER assembler (Pevzner et al. 2001). In this approach, the reads are decomposed into k-mers that in turn become the nodes of a de Bruijn graph. A directed edge between nodes indicates that the k-mers on those nodes occur consecutively in one or more reads. These k-mers take the place of the seeds used for overlap computation in other assemblers (Fig. 2).

Differences between an overlap graph and a de Bruijn graph for assembly. Based on the set of 10 8-bp reads (A), we can build an overlap graph (B) in which each read is a node, and overlaps >5 bp are indicated by directed edges. Transitive overlaps, which are implied by other longer overlaps, are shown as dotted edges. In a de Bruin graph (C), a node is created for every k-mer in all the reads; here the k-mer size is 3. Edges are drawn between every pair of successive k-mers in a read, where the k-mers overlap by k − 1 bases. In both approaches, repeat sequences create a fork in the graph. Note here we have only considered the forward orientation of each sequence to simplify the figure.
Unambiguous stretches of sequence form nonbranching paths in the de Bruijn graph, making it easy to “read off” contigs by walking these paths. Overlaps between reads are implicitly captured by the graph, rather than explicitly computed, saving a substantial amount of computing time. Similar to the overlap graph approach, all copies of a repeat will initially be represented by a single high-coverage node. Repeat boundaries and sequencing errors show up as branch points in the graph, and complex repeats appears as densely connected “tangles.”
Sequencing error complicates the de Bruijn graph, but many errors are easily recognized by their structure in the graph. For example, errors at the end of a read usually create k-mers that occur only once, and therefore form dead-end “tips” in the graph. Errors in the middle of a read create alternate paths called “bubbles” that terminate at the same node. De Bruijn graph assemblers search for these localized graph structures in an error correction phase and remove the error nodes and other low coverage nodes. Mate-pair information can be used to resolve ambiguity, using the coverage at each node to identify repeats, and by searching for unique paths through the graph consistent with the mate pairs.
The main drawback to the de Bruijn approach is the loss of information caused by decomposing a read into a path of k-mers. Compared with conventional assemblers, where a read is a single node in the overlap graph, de Bruijn assemblers initially create multiple nodes for each read, and these nodes may not form a linear path once edges from other reads are added. Furthermore, unlike the overlap graph, the de Bruijn graph is not read coherent (Myers 2005), meaning there may be paths through the graph that form a sequence that is not supported by the underlying reads. For example, if the same k-mer occurs in the middle of two reads, but the reads do not otherwise overlap, the corresponding de Bruijn graph for those reads contains a branching node instead of two separate paths. Short repeats of this type can be resolved, but they require additional processing and therefore additional time.
Another potential drawback of the de Bruijn approach is that the de Bruijn graph can require an enormous amount of computer space (random access memory, or RAM). Unlike conventional overlap computations, which can be easily partitioned into multiple jobs with distinct batches of reads, the construction and analysis of a de Bruijn graph is not easily parallelized. As a result, de Bruijn assemblers such as Velvet and ALLPATHS, which have been used successfully on bacterial genomes, do not scale to large genomes. For a human-sized genome, these programs would require several terabytes of RAM to store their de Bruijn graphs, which is far more memory than is available on most computers.
To date, only two de Bruijn graph assemblers have been shown to have the ability to assemble a mammalian-sized genome. ABySS (Simpson et al. 2009) assembled a human genome in 87 h on a cluster of 21 eight-core machines each with 16 GB of RAM (168 cores, 336 GB of RAM total). SOAPdenovo assembled a human genome in 40 h using a single computer with 32 cores and 512 GB of RAM (Li et al. 2010). Although these types of computing resources are not widely available, they are within reach for large-scale scientific centers.
In theory, the size of the de Bruijn graph depends only on the size of the genome, including polymorphic alleles, and should be independent of the number of reads. However, because sequencing errors create their own graph nodes, increasing the number of reads inevitably increases the size of the de Bruijn graph. In the de novo assembly of human from short reads, SOAPdenovo reduced the number of 25-mers from 14.6 billion to 5.0 billion by correcting errors before constructing the de Bruijn graph (Li et al. 2010). Its error correction method first counts the number of occurrences of all k-mers in the reads and replaces any k-mers occurring less than three times with the highest frequency alternative k-mer.
Choice of assembler and sequencing strategy
Only de Bruijn graph assemblers have demonstrated the ability to successfully assemble very short reads (<50 bp). For longer reads (>100 bp), overlap graph assemblers have been quite successful and have a much better track record overall. A de Bruijn graph assembler should function with longer reads as well, but a large difference between the read length and the k-mer length will result in many more branching nodes than in the simplified overlap graph. The precise conditions under which one assembly method is superior to the other remain an open question, and the answer may ultimately depend on the specific assembler and genome characteristics.
As Figure 3 illustrates, there is a direct and dramatic tradeoff among read length, coverage, and expected contig length in a genome assembly. The figure shows the theoretical expected contigs length, based on the Lander-Waterman model (Lander and Waterman 1988), in an assembly where all overlaps have been detected perfectly. This model, which was widely applied for predicting assembly quality in the Sanger sequencing era, predicts that under ideal conditions, 710-bp reads should require 3× coverage to produce 4-kbp average contig sizes, while 30-bp reads would require 28× coverage. In practice, the model is inadequate for modeling very short reads: The figure also shows the actual contig sizes for the dog genome, assembled with 710-bp reads, and the panda genome, assembled with 52-bp reads. The dog assembly tracked closely to the theoretical prediction, while the panda assembly has contig sizes that are many times lower than predicted by the model. The large discrepancy between predicted and observed assembly quality results from the fact that simplifying assumptions in the model are violated by shorter reads, especially the assumption that the genome is free of repeats. As shown in Figure 1, a larger proportion of a genome is repetitive at shorter read lengths, and consequently an assembler will be forced to create many more contig breaks at repeat boundaries.
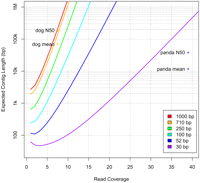
Expected average contig length for a range of different read lengths and coverage values. Also shown are the average contig lengths and N50 lengths for the dog genome, assembled with 710-bp reads, and the panda genome, assembled with reads averaging 52 bp in length.
Further complicating any modeling strategy, second-generation sequencing methods have sequence-dependent coverage biases and nonuniform error rates (Dohm et al. 2008). These sequencing irregularities will cause unexpectedly low coverage regions (e.g., Illumina sequencers have lower coverage in low-GC regions) and consequently more gaps in an assembly. Fortunately, many of these limitations can be overcome by additional oversampling of the genome to boost the low coverage regions.
Figure 3 also shows that even for longer Sanger reads, the theoretical model is a better predictor of N50 contig sizes than of mean contig lengths. An N50 contig size of N means that 50% of the assembled bases are contained in contigs of length N or larger. N50 sizes are often used as a measure of assembly quality because they capture how much of the genome is covered by relatively large contigs.
A good compromise solution to the problem of assembling a genome with short reads is to create a hybrid assembly using a mix of short and long reads. One strategy that we have used with some success is to assemble the short reads with a de Bruijn graph method such as Velvet, and then treat the resulting contigs as reads. The Velvet contigs together with the longer reads can then be assembled with CABOG or another overlap graph assembler.
Another strategy is to assemble the short and long reads using a single de Bruijn graph assembler. In this approach, the long reads are primarily used to disambiguate short repeats. This can work well, although overlap graph assemblers (CABOG and ARACHNE) are more mature than the new short read assemblers and generally produce much better assemblies, especially due to their more sophisticated use of mate pairs. Using an overlap graph assembler with a combination of long and short reads requires that the assembler be carefully tuned to accommodate the shorter reads and potentially higher error rates.
By far the best approach is to use a reference genome sequence, which the assembler will use as a guide to resolve repeats. This is known as comparative assembly (Pop et al. 2004), and the assemblers that can perform this are a special subclass of assemblers. Most human resequencing efforts have followed this approach, if they attempted assembly at all, because it produces a far better result. However, the obvious drawback is that comparative assembly is simply not possible unless the species has already been sequenced and assembled previously. Another drawback is that purely comparative techniques cannot resolve large insertions or structural variations.
Sequencing cost
Any attempt to design a sequencing project must account for cost. Each of the SGS technologies has different costs for library construction and for the sequencing itself. The costs have been changing rapidly and steadily decreasing, but it is worthwhile to present a snapshot of relative costs as they are today. A 2009 study (Wall et al. 2009) examined the costs of transcriptome sequencing with Sanger, 454 Life Sciences (Roche), and Illumina technologies at that time and reported that the lowest cost 454 (GS FLX) method was ∼22 times more expensive, per megabase, than Illumina. Although few other published comparisons are available for the current generation of sequencing machines, unpublished reports indicate that the cost per run is roughly comparable between 454 and Illumina. At a throughput of ∼400 Mb for 454 and 20 Gb for Illumina (and note that these values that are constantly increasing) the 454 technology is ∼50 times more expensive.
Genome coverage and gaps
As coverage increases, the fraction of the genome sequenced increases while the number of gaps decreases. However, each sequencing technology has its own biases that produce gaps in coverage. Conventional Sanger sequencing uses cloning steps that amplify the genome in E. coli, which does not amplify all sequences equally well. SGS technologies avoid cloning in E. coli, but they too seem to have biases. Therefore any genome sequenced with just one technology, regardless of the depth of coverage, is liable to contain gaps due to bias. One way to overcome these biases and to close many gaps is to generate deep coverage in two or more sequencing technologies (Goldberg et al. 2006).
For Sanger sequencing projects, the point of diminishing returns, where additional sequencing yields little additional genomic sequence, falls at ∼8× coverage. For very short reads (<50 bp), higher coverage is clearly necessary, but the optimal depth of coverage has been a rapidly moving target over the past several years. Below, we describe a number of SGS projects that have used different read lengths, depths of coverage, and assembly algorithms, with a mixture of results.
An important side note here is that coverage cannot be computed precisely based on the number of reads generated, because all SGS technologies have a nonnegligible failure rate. This is best illustrated by resequencing projects, in which it is typical to find only 70%–75% of the reads mapping onto the genome. The remaining 25%–30% of the reads fail to map primarily due to low quality. This is not surprising when compared with Sanger sequencing methods from the 1990s, when 20%–25% failure rates were common (Trapnell and Salzberg 2009). Nonetheless, when computing desired coverage, researchers should plan on a yield of ∼70%–75% from the total number of reads generated.
Read length and insert size
In the ideal case, the quality of an assembly will be determined by the read lengths, mate-pair distances, and by the repeat structure of the genome. In general, longer reads make better assemblies because they span more repeats. Similarly, longer insert sizes (mate-pair distances) will increase scaffold sizes, but longer inserts will not always improve contig sizes. For an assembler to close a gap within a scaffold, it must find a set of reads that form an unambiguous path between the flanking contigs. With large gaps, multiple alternative paths through the overlap or de Bruijn graph are much more likely.
For this and other reasons, using a mixture of insert sizes can be very effective. The shortest inserts are used to resolve the small repeats, and longer inserts can resolve progressively longer repeats. In practice, long inserts tend to be less reliable, with a much higher variance in their length distribution.
Published SGS genome assemblies
In this section we survey short-read assembly results that have been published or recently announced. A summary of the de novo short-read assemblies is contained in Table 1, which gives general characteristics of the assemblies. Specific values can vary in how they are computed; for example, the number of contigs depends on the minimum contig length included in the published assembly.
De novo assemblies of second-generation sequencing projects
Human genomes
Initial assembly results with SGS technology consisted primarily of mapping reads to a reference genome. This was the case with several human assemblies, including that of James Watson (Wheeler et al. 2008), which was sequenced with 454 unpaired reads. Genomes from African (Yoruba) (Bentley et al. 2008), Asian (Han) (Wang et al. 2008), and Korean (Kim et al. 2009) individuals were all sequenced with Illumina technology and mapped to the reference human sequence. For the Asian genome, 487 million reads that did not map successfully were assembled using Velvet, but only a small portion of these (0.36%) assembled into contigs of >100 bp. The Korean genome included sequencing of targeted BACs in addition to WGS sequencing.
The above-mentioned African genome data were later assembled de novo to test the ABySS assembler (Simpson et al. 2009). The assembly of the 3.5 billion paired-end reads (lengths 35–46 bp from DNA sequence fragments of ∼210 bp) yielded an astounding 2.76 million contigs with an N50 length of only 1499 bp. These contigs covered only 68% of the human reference genome. The assembly took almost 4 d using a 168-core compute cluster. This same data set was later assembled using SOAPdenovo, which took 40 h on a 32-core 512 GB RAM computer. This latter assembly had an improved N50 contig length of 4.6 kbp and covered 85% of the human reference genome. The current best-published de novo assembly of a human genome was also assembled using SOAPdenovo from a total of 90× coverage of an Asian individual (Li et al. 2010) producing an N50 contig length of 7.4 kbp. These assemblies were computed from older Illumina sequence data (average read length of <40 bp) and would likely improve further using the longer read lengths available today.
Combinations of Sanger and SGS reads
Several large draft genomes have been published that used a combination of Sanger and short-read sequencing. The draft assembly of grapevine (Vitis vinifera, genome size ∼ 500 Mb) reported in (Velasco et al. 2007) combined Sanger and 454 sequencing. An initial assembly of the 6.5× coverage Sanger data was created, and the additional 4.2× coverage of 454 sequence was used to correct errors and fill gaps.
The draft genome sequence of cucumber, Cucumis sativus, was obtained using a combination of Sanger and Illumina sequencing (Huang et al. 2009). Illumina reads represented 68× coverage by pairs from fragment sizes 200, 400, and 2000 bp, while Sanger reads represented coverage of 4× coverage using pairs with insert sizes 2, 4, 6, 40, and 150 kb. Results for three different assemblies—Illumina only, Sanger only, and combined—were reported with the best results obtained, as expected, using the combined data set: N50 contig and scaffold sizes of 19.8 kb and 1.14 Mb, respectively, and totals of 227 Mb in contigs and 244 Mb in scaffolds. It is interesting, however, that although the N50 sizes of the Sanger-only assembly were much smaller (2.6-kb contigs and 19-kb scaffolds), the coverage of the Sanger-only assembly was rather good—204 Mb in contigs and 238 Mb in scaffolds—and better than the Illumina-only assembly (190 Mb in contigs and 200 Mb in scaffolds). The entire genome is estimated to be ∼360 Mb, indicating that something hampered the assembly, possibly a large number of repeats, or problems with the assembler itself, or with the laboratory protocols. The assembly was accomplished using the authors' own software to assemble the Illumina reads first, and then RePS2 (Wang et al. 2002) was used to merge the Illumina scaffolds with the Sanger reads.
Panda
The first de novo, exclusively SGS assembly of a novel, large genome, that of the giant panda, Ailuropoda melanoleura, was recently published by the Beijing Genome Institute (Kohn et al. 2010). This assembly used only Illumina reads averaging 52 bp in length and was done with the SOAPdenovo assembler. Thirty-seven paired-end libraries were constructed, with fragment sizes of 150 bp, 500 bp, 2 kbp, 5 kbp, and 10 kbp, and a total of 218 Genome Analyzer lanes of sequence were generated (not counting 17 lanes discarded due to low quality). This generated roughly 231 Gb of raw sequence (roughly 96× coverage of the 2.4-Gb genome). This was reduced to 176 Gb (73×) of sequence used for all analyses after removing low-quality and duplicate reads—the proportion of duplicate reads ranged from 5% of the 500-bp libraries to 77% of the 10-kb libraries. Further quality filtering of reads generated the 134 Gb (56×) of sequence used in the actual de novo assembly, and of this ∼39× was used to assemble contigs while the remaining data were used as links to create scaffolds. Thus, <60% of the total sequencing data were used in the actual assembly.
The final assembly contained 200,604 contigs (of length at least 100 bp) totaling 2.25 Gb (93.8% of the genome), with impressive N50 contig and scaffold sizes of 36,728 bp and 1.22 Mb, respectively. There were 5201 multicontig scaffolds comprising 124,336 contigs, and a total of 119,135 gaps with mean gap size of only 455 bp. Thus the total span of all contigs and scaffolds (including gaps) was 2.30 Gb, 95.8% of the genome. The remarkably good quality of this assembly is in large part due to the very high depth of sequence coverage, particularly by long-pairs, and the fact that the genome is much less repetitive than primate and rodent genomes.
An interesting comparison is the dog genome, which has a nearly identical genome size (estimated to be 2.45 Gb) and is used for several evolutionary comparisons in the panda paper. The dog genome was assembled at the Broad Institute in 2005 using 7.5× coverage by Sanger sequence data (Lindblad-Toh et al. 2005). The N50 contig size for the dog assembly was 180 kb, and the N50 scaffold size was an impressively large 45 Mb. This rather significant advantage of the dog assembly over the panda assembly is likely due to three factors:
-
Longer Sanger reads: There are many very short gaps in the panda assembly that undoubtedly would be closed by the Sanger reads, which averaged 770 bp long.
-
Longer insert libraries: The sequence available for the dog assembly included 2.2 million reads from a 40-kb fosmid library and 302,000 BAC ends —that cannot be generated by SGS technologies.
-
More mature assembly software: The dog assembly paper reported that improvements to the ARACHNE assembler alone increased contig N50 size from 123 to 180 kb.
It is interesting to note that the panda download site includes several “gene scaffolds,” indicating locations where a gene spans separate scaffolds in the assembly. This information could have been used to combine scaffolds and improve the scaffold N50 value.
Announced but unpublished SGS assemblies
A number of draft SGS assemblies have been announced but have not been published. We describe them here to give a sense of the various strategies currently being used to assemble large genomes.
Cod
An assembly of the cod genome (Gadus morhua, genome size ∼ 800 Mb) (http://www.genomeweb.com/sequencing/norwegian-consortium-assembles-annotates-cod-genome-454-data?page=show) was generated from ∼27× coverage of 454 reads and included paired libraries from 2-, 3-, 8-, and 20-kb fragments. Additional Sanger sequencing of BAC ends was also used to confirm the assembly. The N50 scaffold size is reportedly 571 kb and the scaffolds cover 618 Mb of the genome. The relatively low scaffold coverage and difficulty in accurately estimating the genome size are largely due to the presence of copious repeats in the sequence.
Strawberry
The announced draft assembly of the wild strawberry genome, Fragaria vesca, was obtained using a combination of 454, Illumina and Applied Biosystems SOLiD System sequence data (http://strawberry.vbi.vt.edu). The assembly was created by first using CABOG to assemble the 454 data. Then SOLiD pairs were added to grow scaffolds, using the scaffolder within CABOG. Finally a Velvet assembly of the Illumina data was done, and the contigs were mapped to the 454/SOLiD assembly to fill gaps and correct homopolymer SNP errors. The resulting N50 sizes of contigs and scaffolds were 28 kb and 1.44 Mb, respectively, for this ∼220-Mb genome. There are plans to improve the assembly by incorporating data from a restriction digest of a BAC library.
Turkey
The draft assembly announced for the turkey genome (Meleagris gallopavo, genome size ∼1.1 Gb) was created primarily from a combination of 454 and Illumina sequencing. The 454 sequences included 4 million read pairs from 3-kb and 20-kb fragments plus 13 million unpaired reads. Illumina sequencing included 400 million 74-bp reads from both paired and unpaired sequences. Overall coverage was ∼5× in 454 reads and ∼25× in Illumina reads. Forty thousand Sanger BAC-end sequences, providing ∼6× clone coverage, were also used in the hybrid assembly, which was constructed with the CABOG assembler. The N50 contig and scaffold sizes were 12.6 kb and 1.5 Mb, respectively, with the longest contig being 90 kb and the longest scaffold 9 Mb. These values are substantially smaller than the corresponding ones for the chicken genome, done with Sanger sequencing: N50 contig 36 kb, N50 scaffold 7.1 Mb, longest contig 442 kb, longest scaffold 7.1 Mb. On the other hand, the sequencing costs for turkey were estimated to be <2.5% of those for chicken. It is also interesting that the average sequence coverage in contigs in the turkey assembly was 17×, even though the overall level of sequence coverage was >30×, indicating that this version of the assembly had difficulty incorporating all available sequence data.
Recommendations for SGS sequencing
The above results make it clear that assemblies using SGS reads alone are substantially inferior to what can be accomplished using Sanger sequencing. The two-to-three orders of magnitude cost advantage of SGS, however, will continue to make it much more appealing, and for many genomes it may be the only affordable option. The assembly results now being obtained with SGS sequencing, such as the pioneering panda genome assembly, are scientifically valuable: They cover most of the genome and they produce contigs and scaffolds long enough for comprehensive gene-annotation efforts. These results will continue to improve as SGS read lengths grow, paired-end protocols improve, and assembly software innovations appear.
The keys to good assembly results include deep coverage by reads with lengths longer than common repeats, and paired-end reads from short (0.5–3 kb) and long (>3 kb) DNA fragments. Using currently available sequencing technology, the most cost-effective way to obtain sequence coverage with what are effectively 200–300-bp reads, is to use paired-end Illumina reads from 200–300-bp fragments. With at least 20× coverage in such reads, assemblers using either de Bruijn graphs or overlap graphs should be able to assemble contigs that cover the unique regions of a large genome.
To obtain large scaffolds and fill in repeat-induced gaps, a sequencing project should also generate a large set of reliable paired-end reads. As long as both ends of a pair map uniquely to contigs, the pair can be used for scaffolding, and, to fill in scaffold gaps, we need paired reads in which one read is anchored in a contig and its mate falls in the gap. For this reason, a mixture of several fragment sizes is necessary to resolve the short and long repeats in a genome. Longer (e.g., 454-based) reads are also advantageous in resolving the most complicated repeats, but (potentially modest) improvements to assembly quality may not justify the higher costs of long reads.
More important than the read length of paired reads, however, is the number of distinct, nonchimeric pairs produced. Protocols to generate paired reads are still being refined, and we have seen sequencing runs that suffered from having very few distinct pairs in them, from having numerous redundant pairs (the same pairs occurred repeatedly), and from having chimeric pairs (the paired sequences were not at the expected separation and orientation in the genome). As noted above, the redundancy of the 10-kb library in the panda genome project was 77%, implying that the actual coverage was just under one-fourth of that indicated by the total sequence length. Until paired-end protocols become more effective, sequencing projects will need to identify experienced laboratories that have demonstrated an ability to generate these sequences.
With the assembly software available today, it is technically feasible and cost-effective to build a good assembly entirely from the low-cost short reads produced by Illumina sequencers. Scientists planning such a project should aim to produce relatively deep coverage (30× or more) in paired-end sequences from short DNA fragments (500–1000 bp), and additional coverage (10–20×) in paired ends from longer DNA fragments (3–10 kbp), following a recipe similar to that used in the panda sequencing project. Note that the panda genome project generated just under 100× coverage of raw data, of which only 39× coverage was used after eliminating bad or redundant reads. Coverage requirements can be reduced by using longer reads, but the relationship between coverage and read length is complex. Alternatively, the turkey genome demonstrates that a good assembly can also be achieved using a hybrid strategy that mixes lower coverage (5×) of paired-end 454 sequencing with deeper coverage (25×) Illumina sequencing.
Sequencing technology is a rapidly advancing field, and third-generation sequencing technologies have been announced this year that feature even longer read lengths and insert sizes than were possible with first-generation Sanger sequencing. When these technologies are available, our recommendations and associated cost analyses undoubtedly will change.
Acknowledgments
This work was supported in part by NIH grants R01-LM006845 and R01-GM083873 and by NSF grant IIS-0844494.
Footnotes
-
↵2 Corresponding author.
E-mail salzberg{at}umd.edu.
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.101360.109.
- Copyright © 2010 by Cold Spring Harbor Laboratory Press








