
ABySS: A parallel assembler for short read sequence data
- Jared T. Simpson,1,
- Kim Wong,
- Shaun D. Jackman,
- Jacqueline E. Schein,
- Steven J.M. Jones and
- İnanç Birol,2
Abstract
Widespread adoption of massively parallel deoxyribonucleic acid (DNA) sequencing instruments has prompted the recent development of de novo short read assembly algorithms. A common shortcoming of the available tools is their inability to efficiently assemble vast amounts of data generated from large-scale sequencing projects, such as the sequencing of individual human genomes to catalog natural genetic variation. To address this limitation, we developed ABySS (Assembly By Short Sequences), a parallelized sequence assembler. As a demonstration of the capability of our software, we assembled 3.5 billion paired-end reads from the genome of an African male publicly released by Illumina, Inc. Approximately 2.76 million contigs ≥100 base pairs (bp) in length were created with an N50 size of 1499 bp, representing 68% of the reference human genome. Analysis of these contigs identified polymorphic and novel sequences not present in the human reference assembly, which were validated by alignment to alternate human assemblies and to other primate genomes.
Massively parallel sequencing platforms, such as the Illumina, Inc. Genome Analyzer, Applied Biosystems SOLiD System, and 454 Life Sciences (Roche) GS FLX, have provided an unprecedented increase in DNA sequencing throughput. Currently, these technologies produce high-quality short reads from 25 to 500 bp in length, which is substantially shorter than the capillary-based sequencing technology. However, the total number of base pairs sequenced in a given run is orders of magnitude higher. These two factors introduce a number of new informatics challenges, including the ability to perform de novo assembly of millions or even billions of short reads.
The field of short read de novo assembly developed from pioneering work on de Bruijn graphs by Pevzner et al. (Pevzner and Tang 2001; Pevzner et al. 2001). The de Bruijn graph representation is prevalent in current short read assemblers, with Velvet (Zerbino and Birney 2008), ALLPATHS (Butler et al. 2008), and EULER-SR (Chaisson and Pevzner 2008) all following this approach. As an alternative, a prefix tree-based approach was introduced by Warren et al. (2007) with their early work on SSAKE. This paradigm was also followed in the VCAKE algorithm by Jeck et al. (2007), and in the SHARCGS algorithm by Dohm et al. (2007). On a third branch, Edena (Hernandez et al. 2008) was an adaptation of the traditional overlap-layout-consensus model to short reads.
These short read de novo assemblers are single-threaded applications designed to run on a single processor. However, computation time and memory constraints limit the practical use of these implementations to genomes on the order of a megabase in size. On the other hand, as the next generation sequencing technologies have matured, and read lengths and throughput increase, the application of these technologies to structural analysis of large, complex genomes has become feasible. Notably, the 1000 Genomes Project (www.1000genomes.org) is undertaking the identification and cataloging of human genetic variation by sequencing the genomes of 1000 individuals from a diverse range of populations using short read platforms. Up to this point however, analysis of short read sequences from mammalian-sized genomes has been limited to alignment-based methods (Korbel et al. 2007; Bentley et al. 2008; Campbell et al. 2008; Wheeler et al. 2008) due to the lack of de novo assembly tools able to handle the vast amount of data generated by these projects.
To assemble the very large data sets produced by sequencing individual human genomes, we have developed ABySS (Assembly By Short Sequencing). The primary innovation in ABySS is a distributed representation of a de Bruijn graph, which allows parallel computation of the assembly algorithm across a network of commodity computers. We demonstrate the ability of our assembler to quickly and accurately assemble 3.5 billion short sequence reads generated from whole-genome sequencing of a Yoruban male (NA18507) on the Illumina Genome Analyzer platform.
Results
Algorithmic approach
The ABySS algorithm proceeds in two stages. First, all possible substrings of length k (termed k-mers) are generated from the sequence reads. The k-mer data set is then processed to remove read errors and initial contigs are built. In the second stage, mate-pair information is used to extend contigs by resolving ambiguities in contig overlaps. Details of the assembly algorithm are provided in the Methods section.
Evaluation of ABySS performance
Simulated data
We assembled two sets of simulated, error-free short reads generated from the human reference genome sequence (NCBI Build 36.1) (International Human Genome Sequencing Consortium 2004) to characterize the performance of the algorithm under ideal conditions and to evaluate the frequency of misassembled contigs. Contigs that have an exact, full-length match to the reference genome, or a full-length match with mismatched bases and no alignment gaps, are considered to be correct. Mismatched bases are an artifact of the single nucleotide polymorphism (SNP) removal algorithm, which cannot distinguish sequence “bubbles” formed by heterozygous SNPs in a diploid genome from distinct paralogous regions that differ by one or a few base pairs (see Methods). Since the alternative sequences are retained in a log file, contigs with mismatched bases are not considered to be misassembled. Contigs with more extensive differences from the reference human genome, such as alignment gaps, partial alignments, or split alignments to different chromosomes, are considered to be misassembled.
The first synthetic data set represented all possible error-free 36-mer paired sequences, using a fixed fragment size of 200 bp. We generated simulated reads by sliding a 200 bp window, with a step size of 1 bp, along each chromosome of the reference genome and reporting the first 36 bp and the reverse complement of the last 36 bp. This process produced a data set of perfectly tiled 72-fold read coverage of the reference genome. This data set was assembled using the assembly parameter k = 36 (see Supplemental material) and produced 1.60 million contigs ≥100 bp with an N50 size of 3656 bp. The assembled contigs are highly accurate with 94.4% of the contigs aligning perfectly to the reference human genome and another 5.0% aligning full length with internal mismatches. Together, these contigs represent 80% of the reference genome. The remainder of the genome is represented by contigs <100 bp in size, which correspond to repeat structures that cannot be resolved by the short read paired-end data. In addition, a very small portion of the genome is represented by misassembled contigs (see below). This simulation forms a baseline for the proportion of the reference genome that can be assembled into contigs ≥100 bp given 36 bp paired-end reads from a 200 bp fragment. The assembly statistics are summarized in Table 1.
Assembly statistics for perfectly tiled, fixed 200-bp fragment, error-free simulated data
The second synthetic data set simulated a random sampling of error-free 36-mer paired-end reads. Instead of a fixed fragment size of 200 bp, we applied a fragment size distribution corresponding to the empirical distribution of the experimental data set SRA000271 (see the next section and Supplemental Fig. 1). We sampled the genome to provide an average of 42-fold sequence coverage of the reference genome, again mimicking the experimental data, and assembled using the assembly parameter k = 27. Similar to the assembly of the perfectly tiled simulated data, 95.6% of the contigs ≥100 bp aligned perfectly to the reference genome and 3.8% aligned with internal mismatches. These contigs represent 71% of the reference genome. These assembly statistics are summarized in Table 2.
Assembly statistics for 42× sampled, error-free simulated data
A small number of contig misassemblies are anticipated to arise during the contig merging process because the merging algorithm accommodates imperfect data and a wide distribution of fragment sizes, such as one would expect in experimental data sets. These misassemblies can range in severity from minor, for example, a small indel introduced by incorrectly estimating the copy number of a local repeat (found in 0.5% of contigs for the tiled simulation and 0.5% for the sampled simulation), to major misassemblies where distinct regions of the genome are incorrectly brought together (found in 0.04% and 0.1% of the tiled and sampled simulations, respectively). In total, misassembled contigs represent less than 1% of the total contigs and less than 1% of the genome in both assemblies using error-free data.
Experimental sequence data
We obtained sequence data for the genome of an African male individual (HapMap DNA identifier NA18507) (International HapMap Consortium 2003, 2007) from the NCBI short read archive (accession no. SRA000271). The sequence was generated by Illumina, Inc. using their Genome Analyzer platform (Bentley et al. 2008).
The data set consists of 3.5 billion paired-end tag reads with read lengths ranging from 36 to 42 bp and a median fragment size of ∼210 bp (see Supplemental Fig. 1), representing the human genome with an average of 42-fold sequence redundancy. An initial quality check of the data was performed by aligning the sequences to the human reference genome using the MAQ aligner (Li et al. 2008). Analysis of the alignments revealed that 72% of the aligning reads perfectly matched the human reference. The per-base error rate was estimated to be ∼1.4% based on the number of mismatches in the alignments, however, this includes mismatches that represent polymorphic sites.
The genome assembly was performed using the assembly parameter k = 27 (see Supplemental material). The first phase of the assembly, which does not use the paired-end information, required 15 h to complete. An additional 3 d were required to merge contigs using the paired-end information (see Supplemental material for a description of the cluster architecture). After merging with paired-end data, there were 2.76 million contigs ≥100 bp in size, with an N50 size of 1499 bp (Table 3).
Assembly statistics for data from the NA18507 Yoruba individual
Of these 2.76 million contigs, 94.2% were assembled correctly, with full-length alignments to the reference genome, a minimum of 95% sequence identity, and alignment gaps no greater than 50 kb (see Methods). An additional 4.6% of the contigs align to the reference genome with at most four unmatched bases at contig termini. Unmatched bases at the termini occur where sequence errors are undetected due to poor sequence coverage; therefore, we also consider these contigs to be correct. Together, 98.8% of the contigs are deemed correct, covering 68.2% of the human reference sequence. These figures are only slightly lower than the results obtained from the stochastic 42× simulation. When the restriction on contig size is removed, we observe that 90% of the reference genome is covered by contigs (see Supplemental Fig. 2). If we only consider uniquely aligning contigs, 81% of the reference genome is covered.
For a small portion of the 2.76 million contigs (0.08%) we observe significant alignment inconsistencies, where contigs have either both ends aligning to different chromosomes, ends aligning to the same chromosome but on the opposite strand, or with ends aligning over 50 kb apart; these may represent contigs crossing breakpoints of translocations, inversions, or large deletions, respectively. However, the observed frequency of such contigs is within range of the 0.04% and 0.10% of contigs with these types of anomalous alignments that we observed in the assemblies with simulated perfect-coverage data and simulated sampled data, respectively, indicating that the majority of these are likely to be incorrectly assembled contigs.
Polymorphic and novel human sequence identified in the assembled experimental data
Analysis of the alignments of correctly assembled contigs to the NCBI human reference genome revealed 110,177 deletions and 101,578 insertions (see Fig. 1). The distribution of deletion sizes has a prominent peak in the 280–350 bp range, which has been previously observed in other human genome sequencing projects (Levy et al. 2007; Bentley et al. 2008; Wheeler et al. 2008). The peak represents 545 deletions, 95% (517) of which overlap with Alu insertions in the human reference genome. Approximately 24% of these (125) have not been identified as retrotransposon insertion polymorphisms (RIPS) in dbRIP (Wang et al. 2006). In addition, we have identified 23 sites at which ∼6 kb known polymorphic L1 retrotransposons are absent in the genome of the Yoruban male. Structural variation in this individual relative to the reference genome has been previously identified (Redon et al. 2006; Bentley et al. 2008; Kidd et al. 2008); however, comparison with this data is beyond the scope of this paper.
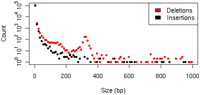
Distribution of insertion and deletion sizes, up to 1000 bp, plotted using a logarithmic scale. There is a pronounced deletion peak around 320 bp, which corresponds to the Alu family of retrotransposons.
Approximately 2% of the contigs assembled by ABySS, consisting of 32,577 contigs totaling 22.4 Mb, aligned only partially or not at all to the NCBI human reference genome. We evaluated alignments of these “orphaned” contigs to the HuRef (Levy et al. 2007) and Celera (Venter et al. 2001) human assemblies. Of these 32,577 contigs, 9208 comprising 5.8 Mb of sequence align full length or near full length (with at most four unmatched bases at contig termini) to HuRef (Levy et al. 2007) (NCBI accession nos. AC000133–AC000156). An additional 1529 contigs (664 kb) aligned to the Celera assembly (Venter et al. 2001) (NCBI accession nos. AC000044–AC000068) or to unassigned Celera contigs. These 10,737 contigs, representing known human sequence not present in the NCBI human reference genome, bring the total percentage of contigs considered to be correctly assembled to 99.4%.
We attempted to validate the remaining contigs by alignment to the chimpanzee genome. Of the remaining 21,840 contigs, we identified 3431 contigs totaling over 1.8 Mb that align completely or near full length to the chimpanzee reference genome (Chimpanzee Sequencing and Analysis Consortium 2005) with at least 95% identity. The majority of these contigs (2231 contigs representing 750 kb) aligns partially or poorly to the human genome and aligns to nonorthologous regions in the chimpanzee genome, and we were therefore unable to identify the genomic location of these novel human sequences. For 1200 contigs with a partial alignment to the human genome and alignment to an orthologous region in the chimpanzee genome we attempted to identify the insertion sites (see Methods). We were able to identify precise insertion sites from 246 such contigs, two of which walked into a sequence gap in the NCBI human reference assembly. Approximately one-third of the remaining 244 insertion sites occur within a larger region of known structural variation annotated in the Database of Genomic Variants (Iafrate et al. 2004), and almost half of these are known to include an insertion.
Another 1725 contigs, encompassing 630 kb, appear to contain novel sequence, since no high-quality alignment to the three human assemblies or the chimpanzee genome could be determined. Only 22% of these could be aligned (with a minimum 90% identity spanning at least 50% of the contig) to either the orangutan genome (ftp://genome.wustl.edu/pub/organism/Primates/Pongo_pygmaeus_abelii) or the rhesus macaque genome (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007). The remaining 16,684 contigs have partial alignments to the human or chimpanzee genomes, but do not meet our criteria as correctly assembled contigs.
Comparison of short read assemblers
To provide context to the performance of ABySS, we performed a comparison with previously published short read assemblers. We used a data set consisting of 20.8 million paired-end 36 bp Illumina reads from a 200 bp insert E. coli library (NCBI Short Read Archive, accession no. SRX000429). We performed assemblies with ABySS, Velvet (Zerbino and Birney 2008), EULER-SR (Chaisson and Pevzner 2008), SSAKE (Warren et al. 2007), and Edena (Hernandez et al. 2008). All assemblers were run in paired-end mode with the exception of Edena, which does not support the use of paired-end information in contig construction. Velvet generates scaffolds by joining contigs with a series of “N” bases. The scaffolds were split at these junctions into their constituent contigs for analysis. Contigs aligning to the reference genome with fewer than five consecutive base mismatches at the termini and at least 95% identity were considered to be correct. A summary of the assembly comparison is presented in Table 4. All the assemblers were able to accurately reconstruct the majority of the E. coli genome with contigs ≥100 bp. However, there is a wide range in terms of contig size and accuracy. ABySSs performance is competitive with the other short read assemblers.
Comparison of assemblies of E. coli K12 MG1655 short read data
Discussion
We report here on ABySS, a parallel sequence assembler. With the novel distributed de Bruijn graph approach in ABySS, we are able to parallelize the assembly of billions of short reads over a cluster of commodity hardware. This method allows us to cost effectively increase the amount of memory available to the assembly process, which can scale up to handle genomes of virtually any size. We have used ABySS to assemble billions of short reads from a human resequencing project. While our initial results are promising, the field of de novo assembly of short reads—especially from mammalian genomes—is still rapidly developing. Improvements to both the underlying sequencing platforms and the assembly algorithms will help increase the quality and utility of the resulting assemblies. The next generation sequencing platforms are continuing to achieve longer read lengths. This will allow a larger k-mer size to be used, which will improve the assemblies by reducing the number of spurious overlaps and consequently decreasing the complexity of the de Bruijn graph. Paired-end sequencing from longer inserts will be critical to increasing the contiguity of mammalian assemblies as long repeat regions are a significant barrier to contig growth. New assembly techniques, such as a hybrid assembly using the high coverage read depth provided by the Illumina platform with the longer GS FLX reads, is also a promising avenue for improving the contiguity of the assembly and resolving repetitive regions.
Unlike alignment-based approaches for analyzing short reads, a de novo assembly allows direct identification and precise localization of insertions and deletions relative to the human reference genome sequence, in addition to identifying novel sequence. From our short read assembly of a single individual, we have identified ∼8.9 Mb of sequence not represented in the human reference assembly, 2.4 Mb of which is also not present in the alternate human assemblies. We believe this unbiased approach will lead to substantial insights into the variation present in human genomes, especially in cases where significant variation is anticipated, such as tumor genomes. ABySS will be particularly useful when sequencing organisms for which no reference sequence is available.
Methods
Overview
A de Bruijn graph data structure, as first proposed by Pevzner et al. (2001) and subsequently refined by Chaisson and Pevzner (2008), and Zerbino and Birney (2008), is used as the basis of our assembler. A de Bruijn graph is a directed graph that compactly represents a homogeneous overlap between sequences. The de Bruijn graph is constructed by creating a vertex for every sequence of length k (referred to as a k-mer) in the data set and joining vertices with an edge when they overlap by k-1 bases. The assembly process can then be seen as merging nodes of the graph when they are unambiguously connected. Prior to the concatenation of nodes however, the graph must be cleaned of vertices and edges created by sequencing errors. We have developed a similar process to EULER-SR (Chaisson and Pevzner 2008), Velvet (Zerbino and Birney 2008), and Edena (Hernandez et al. 2008) to handle read errors. First, short “dead-end” branches are removed by the algorithm. More complex errors that form small, divergent bubbles in the graph are then removed and recorded in a subsequent step. We iterate over these two error removal steps to correct read errors that are in close proximity. We have parallelized these processes to allow our assembler to efficiently scale with genome size.
Distributed de Bruijn graph
At the core of the assembly algorithm is our unique representation of a de Bruijn graph. In this representation, adjacent sequences need not be physically located on the same computer, allowing us to distribute the sequences over a cluster of computer nodes. To distribute the de Bruijn graph over a network of computers we need to address two issues. First, the location of a given k-mer must be deterministically and efficiently computable from the sequence of the k-mer. Second, the adjacency information between k-mers must be stored in a manner that is independent of the actual location of the k-mer.
The location of each k-mer is computed with a simple hashing function. A numerical value {0, 1, 2, 3} is assigned to bases {A, C, G, T} to calculate the base-4 representation of a given k-mer. A hash value is then computed from this numerical value. We apply the same procedure on the reverse complement of the sequence, and combine the two values by the XOR operation on their bit representations. This value, modulus the number of nodes, K, is the index used to assign the k-mer to one of the nodes. Since the XOR operation is commutative, the assignment is invariant under reverse complementation. To take advantage of this distributed representation, it is desirable to evenly distribute the set of all possible k-mers over the number of available nodes, to the extent possible by the hash function used.
To store the adjacency information between k-mers we have developed a compact representation of the edges. A single k-mer, or vertex, can have up to eight edges—one for every possible one-base extension, {A, C, G, T}, in either direction. This information can be efficiently stored in 8 bits per k-mer, where one bit represents the presence or absence of each edge. The adjacent k-mers are easily generated from this information and their cluster locations can be deterministically computed by the method described above.
Implementation
ABySS is implemented in C++ and uses the MPI (Message Passing Interface) protocol for communication between nodes. For the internal hash tables, the Google sparse hash library (http://code.google.com/p/google-sparsehash/) is used. Sequences are hashed using the function developed by B. Jenkins (http://burtleburtle.net/bob/c/lookup3.c). The latency and bandwidth of the cluster network can have a significant impact on the performance of a parallel application and require special consideration. To mitigate the latency of the communications link, a nonblocking communications model is used. Each communications message is given a unique identifier. The sending process does not wait for an immediate response, but rather saves the current state of the operation, keyed by the message ID, and continues to process other operations. When a response is received for a particular message, the saved state information is retrieved by using the message ID and the original task continues. This system allows many simultaneous operations on each cluster node, effectively hiding the latency of the network link. As the messages passed between cluster nodes tend to be very short, the messages are collected into larger, 1 kB packets to minimize the impact of communication overhead.
Assembly algorithm
The assembly is performed in two major steps. First, without using the paired-end information, contigs are extended until either they cannot be unambiguously extended or come to a blunt end due to a lack of coverage. In the second step the paired-end information is used to resolve ambiguities and merge contigs.
Building the graph
The data are first loaded into the distributed de Bruijn graph, during which any sequences with unknown bases (“N” or “.” for the Illumina reads) are discarded. Each input sequence l-mer is broken into (l − k + 1) overlapping k-mers by sliding a window of length k along the input sequence. The cluster node index of the k-mer is computed and the k-mer is assigned to this node for storage in a hash table. As a given sequence and its reverse complement are considered to be equivalent, a sequence is not added to the hash table if the reverse complement of the sequence is already present.
Once the k-mers have been loaded into the distributed de Bruijn graph, the adjacency of the k-mers is computed. For each k-mer in the sequence collection a message is sent to its eight possible neighbors. If the neighbor exists there must be a k-1 overlap with the originating k-mer and the adjacency information is set accordingly.
Read errors
Before merging vertices into contigs, the graph must be cleaned of vertices and edges created by sequencing errors. The most prevalent structure caused by sequencing errors is a “dead-end” branch, formed by reads that are a mixture of correct and incorrect k-mers. The correct k-mers of a read connect the incorrect k-mers of the read to the canonical portion of the graph. As incorrect sequences are likely to be unique and most will not have an extension, one end of the branch will terminate with no extension (see Supplemental Fig. 3). To eliminate these structures, branches that contain a dead-end are identified, traced backward until a point of ambiguity is reached, and if the branch is shorter than a threshold length, it is removed from the graph. This process is applied iteratively, increasing the threshold length at each step to remove longer branches that were uncovered by the removal of shorter branches. The branch removal algorithm is sensitive to the choice of the k-mer parameter. If the k-mer parameter is too high, the sequence graph will be broken in many places and it will be difficult to determine if a dead-end branch arises from a read error or from a lack of k-mer coverage. In the latter case, a correct sequence may be removed from the graph resulting in shorter contigs. For further information regarding the choice of the k-mer parameter, see the Supplemental material.
In rare cases, coincident read errors can cause a false branch that is joined on both sides to the canonical portion of the de Bruijn graph. These cases appear in the graph as a path divergence at a single location that converges after k nodes (referred to as a bubble; see Supplemental Fig. 4). This structure also occurs as a result of single nucleotide differences representing allelic variation in a diploid genome or nearly perfect duplicated sequences. By removing such bubbles, contigs can be unambiguously extended further. To remove the bubbles, each point of divergence is found in the graph. Each path from the point of divergence is traced forward looking for the paths to join after n nodes, where k ≤ n ≤ 2k. If the paths join, the path with lower read coverage is removed and all the paths are stored in a log file.
Bubbles in the sequence graph can also form from highly similar repetitive regions in the genome. In these cases the bubble removal algorithm will simplify the repeat to a single sequence. This effect can be seen in the perfect simulation where ∼5% of contigs contained mismatched bases resulting from oversimplification of the graph. As the removed path is stored in a log file, it is possible to recover this information after the assembly is complete.
Vertex merging
The final step in the first phase of the assembly is to merge vertices linked by unambiguous edges. To do this, ambiguous edges are removed from the graph, and the vertices are then merged by the remaining unambiguous edges, creating the initial contigs.
Contig merging using paired-end information
The second phase of the assembly uses the paired-end information, if available, to resolve ambiguities between contigs. The paired-end information is used to identify contigs that can be linked together. The reads are aligned to the initial contigs to create a set of linked contigs, which is filtered to remove erroneous links caused by mispaired or misaligned reads. Two contigs are considered to be linked if at least p pairs (by default p = 5) join the contigs. For each contig Ci, the set of contigs Pi is generated from the list of contigs that are paired to Ci. A graph search is then performed to look for a single unique path (a sequence of contigs) from Ci through the de Bruijn graph, that visits each contig in Pi. As the de Bruijn graph can be extremely dense in repetitive areas, we use a heuristic rule to limit the number of vertices visited in the search and hence keep an upper limit on the computational cost to perform this search. Prior to the search, we infer a distance between Ci and each contig in Pi using a maximum likelihood estimator based on the read pairs aligned to the contigs (see Supplemental material). By constraining the search by these distance estimates, we can cull entire branches of the search tree when it can no longer yield a valid solution. This process is repeated for each contig Ci and the final step stitches together the consistent paths to generate the contigs of the final assembly.
Assembly analysis
For all data sets, only contigs ≥100 bp in length were evaluated. Contigs aligning to the reference genome with fewer than five consecutive base mismatches at the termini and at least 95% identity are considered to be correct, except in the case where an alignment contains a gap greater than 50 kb.
The contigs from the simulated data set were aligned to the reference human genome (NCBI Build 36.1). Contigs assembled from experimental data were initially screened for Epstein-Barr virus (EBV) genomic sequences, which were used to establish the NA18507 cell line, using a BLASTN (Zhang et al. 2000) search against a local database consisting of two EBV strains (GenBank accession nos. AJ507799 and DQ279927) (Deininger et al. 1982; Dolan et al. 2006). The remaining contigs were then aligned to the human reference genome (NCBI Build 36.1) using Exonerate 2.0.0 (Slater and Birney 2005). These contigs were also aligned to HuRef (Levy et al. 2007) and the chimpanzee genome (Chimpanzee Sequencing and Analysis Consortium 2005) (sequence downloaded from UCSC; http://hgdownload.cse.ucsc.edu/downloads.html#chimp panTro2 assembly). Contigs that did not align to the reference human genome were aligned to the Celera assembly (Venter et al. 2001). The Celera assembly is composed of DNA sequences from five individuals, primarily from J.C. Venter (Levy et al. 2007). Although HuRef is an improvement of the Celera genome in terms of the number of sequence gaps and bases assembled overall (Levy et al. 2007), HuRef represents a single individual and thus we are still able to identify contigs that align to the Celera assembly and not HuRef. The NCBI Blast server has additional unassigned sequences from Celera. The contigs were aligned by BLAST using “blastcl3” (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/LATEST/), a BLAST client.
The UCSC “liftOver” tool (Karolchik et al. 2008) was used to map the human coordinates of partially or poorly aligning contigs to orthologous regions in the chimp genome (McConkey 2004). If a contig aligns full length or near full length to the chimpanzee genome, and the converted coordinates of the alignment to the human genome were found to map within this orthologous region, the contig was considered to contain a sequence insertion site. Where the span of the alignment to chimpanzee was >20 bp longer than the alignment to the human reference genome (true for 246 contigs) we were able to identify the location of the insertion site. This was determined from the human genome alignment by taking the coordinate proximal to the unaligned novel sequence. For 858 contigs, the difference in the span of the alignments between human and chimp is ≤20 bp, and for 96 contigs we were unable to determine the precise insertion point due to difficulty in converting alignment coordinates between the human reference and the chimpanzee reference.
Structural variation
Insertions and deletions were identified from alignment of the assembled contigs to the human reference genome (NCBI Build 36.1). Only contigs considered correct (as outlined in Assembly Analysis above) were used. Insertions and deletions were identified by parsing gapped Exonerate alignments. In some cases, Exonerate alignments are split into two alignments when an alignment gap is flanked by the exact sequence at either end. These broken alignments were “stitched” together to form a single alignment with a large gap, allowing us to identify contigs with large insertions or deletions.
Escherichia coli K12 assembly and analysis
E. coli K12 substrain MG1655 Illumina reads were downloaded from the NCBI short read archive (accession no. SRX000429) and assembled with ABySS, Velvet, SSAKE, EULER-SR, and Edena. For each assembler, the assembly parameters were tuned to provide the highest value of N50. The version number for each assembler, and the parameters used in the assembly, are given in the Supplemental material. The contigs from the assemblies were aligned to the E. coli K12 MG1655 reference genome (RefSeq accession no. NC_000913). The criteria stated above in Assembly Analysis were used to evaluate the contigs from various assemblers and classify a contig as correct. Since rearrangements are not expected, contigs with broken alignments were not considered to be correct in this case. Genomic coverage was calculated from full-length, partial, and broken alignments with at least 95% identity to the reference genome. Contigs that aligned with less than 95% identity were considered to be incorrect.
Additional methods
Further details of the assembly algorithm and analysis can be found in the Supplemental material.
Software availability
Software binaries and instructions are available at http://www.bcgsc.ca/platform/bioinfo/software/abyss.
Acknowledgments
We thank M.A. Marra for his support, and Illumina, Inc. for publicly releasing the SRA000271 sequence data. The draft Pongo pygmaeus abelii sequence assembly was provided by the Genome Sequencing Center at Washington University School of Medicine in St. Louis. S.J.M.J. is a senior scholar of the Michael Smith Foundation for Health Research. Funding was provided by Genome Canada, Genome British Columbia, and the British Columbia Cancer Foundation.
Footnotes
-
↵1 Present address: Wellcome Trust Sanger Institute, Hinxton, Cambridge CB10 1SA, United Kingdom.
-
↵2 Corresponding author.
E-mail ibirol{at}bcgsc.ca; fax (604) 876-3561.
-
[Supplemental material is available online at www.genome.org. Software binaries and instructions are available at http://www.bcgsc.ca/platform/bioinfo/software/abyss.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.089532.108.
-
- Received November 26, 2008.
- Accepted February 24, 2009.
-
Freely available online through the Genome Research Open Access option.
- Copyright © 2009 by Cold Spring Harbor Laboratory Press








