
Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses
- Besma Ben Amor1,6,
- Sonia Wirth1,6,7,
- Francisco Merchan1,
- Philippe Laporte1,
- Yves d’Aubenton-Carafa2,
- Judith Hirsch3,8,
- Alexis Maizel1,
- Allison Mallory5,
- Antoine Lucas2,9,
- Jean Marc Deragon4,
- Herve Vaucheret5,
- Claude Thermes2 and
- Martin Crespi1,10
- 1 Institut des Sciences du Végétal (ISV), CNRS, 91198 Gif-sur-Yvette, France;
- 2 Centre de Génétique Moléculaire (CGM), CNRS, 91198 Gif-sur-Yvette, France;
- 3 Biomedal S.L., 41092 Sevilla, Spain;
- 4 Université de Perpignan Via Domitia, CNRS UMR5096 LGDP, 66860 Perpignan Cedex, France;
- 5 INRA Laboratoire de Biologie Cellulaire, 78026 Versailles Cedex, France
-
↵6 These authors contributed equally to this work.
Abstract
Long non-protein coding RNAs (npcRNA) represent an emerging class of riboregulators, which either act directly in this long form or are processed to shorter miRNA and siRNA. Genome-wide bioinformatic analysis of full-length cDNA databases identified 76 Arabidopsis npcRNAs. Fourteen npcRNAs were antisense to protein-coding mRNAs, suggesting cis-regulatory roles. Numerous 24-nt siRNA matched to five different npcRNAs, suggesting that these npcRNAs are precursors of this type of siRNA. Expression analyses of the 76 npcRNAs identified a novel npcRNA that accumulates in a dcl1 mutant but does not appear to produce trans-acting siRNA or miRNA. Additionally, another npcRNA was the precursor of miR869 and shown to be up-regulated in dcl4 but not in dcl1 mutants, indicative of a young miRNA gene. Abiotic stress altered the accumulation of 22 npcRNAs among the 76, a fraction significantly higher than that observed for the RNA binding protein-coding fraction of the transcriptome. Overexpression analyses in Arabidopsis identified two npcRNAs as regulators of root growth during salt stress and leaf morphology, respectively. Hence, together with small RNAs, long npcRNAs encompass a sensitive component of the transcriptome that have diverse roles during growth and differentiation.
Non-protein coding RNAs (npcRNAs) are a class of RNAs that do not encode proteins, but instead their function lies on the RNA molecule. They are a heterogeneous group and have been divided into different classes according to their length and function. With respect to length, npcRNAs can range from 20 to 27 nucleotides (nt) for the families of microRNAs (miRNAs) and small interfering RNAs (siRNAs), 20–300 nt for small RNAs commonly found as transcriptional and translational regulators, or up to and beyond 10,000 nt for medium and large RNAs involved in other processes, including splicing, gene inactivation, and translation (Costa 2007). We use the term non-protein-coding RNAs instead of noncoding RNAs as every sequence has the potential to be coding, and certain large npcRNAs might encode small oligopeptides, which could be translated under specific conditions as shown for a pentapeptide located inside rRNA, a canonical RNA in Escherichia coli (Tenson et al. 1996). In recent years, numerous novel npcRNA candidates have been identified in a variety of organisms from E. coli to Homo sapiens (Argaman et al. 2001; Storz et al. 2004; Washietl et al. 2005).
Several strategies have been employed to detect and discover novel npcRNAs, including both experimental and computational screenings (Huttenhofer et al. 2002). Genomic approaches, such as tiling arrays and systematic sequencing of full-length cDNA libraries, in model organisms have recently revealed that much larger portions of eukaryote transcriptomes represent non-protein-coding transcripts than previously believed (Okazaki et al. 2002; Numata et al. 2003; Rinn et al. 2003; Ota et al. 2004; Chekanova et al. 2007). Diverse npcRNAs, including a surprising number of antisense RNA transcripts, pseudogenes, and truncated transcripts, have been described (Prasanth and Spector 2007). Certain npcRNAs, referred to as riboregulators, control the stability or translation of specific mRNAs and, in this way, regulate developmental events or stress responses in eukaryotic cells (Erdmann et al. 2001). As very few riboregulators involved in development were previously revealed by classical genetic approaches, it has been proposed that riboregulators may fine-tune mRNA levels in the cell and play a more critical role in the adaptation of developmental processes rather than in differentiation per se.
The most well-studied npcRNA species are single-stranded, 20- to 27-nt small RNAs belonging to two classes, miRNAs and siRNAs, both known to have essential roles in the four eukaryote kingdoms (protists, fungi, plants, and animals). In plants, miRNAs and siRNAs differ in their biogenesis, but both function by guiding target mRNA cleavage after integration into a ribonucleoprotein complex: the RISC (RNA-induced silencing complex) invariably containing a member of the AGO protein family (Vaucheret 2006). In contrast, most animal miRNAs appear to repress translation (Chapman and Carrington 2007). The miRNAs are single-stranded, 21-nt RNA molecules deriving from partially-complementary RNA precursors, which are mainly transcribed by RNA polymerase II from intergenic regions, although few miRNA genes are located in introns of protein-coding genes. It has been estimated that miRNA genes could represent more than 1% of the expressed genome in worms and humans where it has been proposed that a single miRNA could regulate at least 100 mRNA targets (Lim et al. 2005), underlining the relevance of this post-transcriptional regulatory mechanism. In Arabidopsis, 118 putative miRNA loci have been identified (Jones-Rhoades et al. 2006), which represent 42 families, 20 families being conserved in other species such as rice (Bonnet et al. 2004; Sunkar et al. 2005) or poplar (Lu et al. 2005). DICER-LIKE 1 (DCL1) is the main enzyme responsible for mature miRNA production, although DCL4 was shown to process at least two young miRNA genes (Rajagopalan et al. 2006).
In plants, in addition to miRNAs, there exists a great diversity of siRNA: the heterochromatic siRNAs (hc-siRNAs), the trans-acting siRNAs (tasiRNAs), and the natural antisense siRNAs (nat-siRNAs). The common trait during the biogenesis of the first two siRNA species is the generation of a fully complementary double-stranded RNA (dsRNA) by the action of an RNA-dependent RNA polymerase (RDR) (Vaucheret 2006). The highly diverse 24-nt hc-siRNAs are produced through the action of DCL3 and RDR2 and are linked to the formation of heterochromatin. The tasiRNAs derive from large npcRNAs that are targets of miRNAs. This miRNA-dependent processing generates shorter npcRNA molecules that are targeted by RDR6 to produce dsRNA that are processed by DCL4 to generate 21-nt tasiRNAs that integrate into RISC complexes. In this way, novel tasiRNAs are generated from the action of nonhomologous miRNAs, amplifying the diversity of small RNA regulated targets (Vaucheret 2006). The nat-siRNAs are generated from a pair of genes transcribed in antisense orientation generating natural double-strand transcripts. They have been identified in Arabidopsis plants growing under high NaCl conditions where one overlapping gene is constitutively expressed and the other induced by salt stress. The resulting dsRNA molecules are cleaved by DCL2 and DCL1 enzymes to generate the nat-siRNA, which cleaves the mRNA of the constitutive gene and leads to salt tolerance (Borsani et al. 2005).
In contrast to small RNAs, much less is known about the large and diverse population of long npcRNAs. This heterogeneous class of transcripts generally does not contain any long open reading frame (ORF) (e.g., no ORF coding more than 70 amino acids). Even though some of these genes may encode oligopeptides, it has been shown that their RNA moiety plays a critical role in their function. In order to achieve their function, they interact with proteins to regulate transcription, translation or mRNA stability (Yamashita et al. 1998; Campalans et al. 2004; Prasanth and Spector 2007; Filipowicz et al. 2008; Sasidharan and Gerstein 2008). Furthermore, several of these npcRNAs are precursors of miRNAs and tasiRNAs (Reinhart et al. 2002; Hirsch et al. 2006). Like some miRNAs, certain npcRNAs are induced in various developmental processes as well as during abiotic stress responses in plants and animals (Jones-Rhoades et al. 2006; Mendes Soares and Valcarcel 2006; Prasanth and Spector 2007; Sunkar et al. 2007). In Caenorhabditis elegans, 25 npcRNAs are regulated in seven developmental stages and two stimulated conditions (He et al. 2006). We have identified 15 plant npcRNAs displaying diverse tissue-specific expression patterns and/or regulation by environmental stimuli (Hirsch et al. 2006).
In this work, we performed a genome-wide in silico screen of a full-length Arabidopsis cDNA library (Castelli et al. 2004) and identified 33 npcRNAs, including 13 that are antisense to protein-coding transcripts. Analyses of these new npcRNAs together with the 43 npcRNAs that we reported previously revealed that 34 potentially give rise to small RNAs, and consistently, we identified several novel mi/siRNA precursors from this group. A new npcRNA accumulated in dcl1 mutants without forming known mi/siRNAs was identified, while abiotic stresses regulated the expression of 22 npcRNAs. Based on their expression profiles, stable secondary structures, and/or sequence homologies, we selected 12 npcRNAs for functional studies in Arabidopsis plants. Overexpression of two of these npcRNAs revealed roles in leaf differentiation and salt stress responses, respectively, suggesting that at least a subset of these newly identified npcRNAs has roles in developmental or stress adaptation programs.
Results
Identification of new Arabidopsis npcRNAs
A number of npcRNAs detected in Arabidopsis databases could be truncated mRNAs, and as such, further analyses are required to identify bona fide npcRNAs. Our previous (Hirsch et al. 2006) bioinformatic analyses, which filtered 172,495 expressed sequence tags (ESTs) and 24,985 mRNAs retrieved from NCBI databases, identified 43 npcRNA genes that shared certain functional elements, such as a high GC content, an atypical compositional skew (over-representation of T versus A) and the presence, in certain cases, of significantly stable RNA secondary structures. Using a similar approach, we analyzed a collection of 18,000 full-length cDNAs (Castelli et al. 2004) and identified 22 additional candidate npcRNA genes, as well as another 11 npcRNAs that are antisense to annotated genes with an overlapping region of at least 50 bp (for the 33 new npcRNAs and their names, see Supplemental Table 1). Our 43 previously identified npcRNAs also were identified by our new analysis, although in certain cases, we introduced small changes in their annotations; notably three were antisense to protein-coding mRNAs. The size of these 33 new npcRNAs ranges from 265–1879 nt with a mean of 1013 nt, and they harbor one or several introns in one third of the cases. We looked for potential ORFs in the npcRNA, i.e., segments of RNA that, between a start and a stop codon, are multiples of 3 nt. In 22 of them, the longest ORF (mean, 100 nt) is preceded by two to 30 ATG codons, rendering ribosome initiation at this ORF unlikely (Supplemental Table 1). The majority of the selected npcRNAs displays a high GC content (mean GC%, 38%) and a significant abundance of T over A (skew, S already observed for the previous set of npcRNAs, calculated as in Hirsch et al. 2006). Nineteen out of the 22 npcRNAs without an annotated antisense partner display a positive T/A skew, further reinforcing their strand-specific transcription (Touchon et al. 2004). Highly significant Z-scores (Z-score > 5) (Hirsch et al. 2006) suggest the presence of stable RNA secondary structures for five of the new putative npcRNAs (Supplemental Fig. 1). BLASTN analysis revealed that three npcRNAs show significant nucleotide similarities independent of their encoded ORFs, whereas three contain conserved small ORFs (sORFs) (Supplemental Table 1). When the sORF encoded in an npcRNA is much smaller than the similar protein, these genes may correspond to pseudogenes (three genes, indicated as “pp” in Supplemental Table 1). Apart from antisense genes, we could not find any evidence of preferential silent codon nucleotide substitutions. Finally, npc511, npc513, npc520, npc530, and npc541 contain at least one 100-nt box (conserved greater than 90%) in another region of the Arabidopsis thaliana genome.
Nine npcRNAs correspond to miRNA, tasiRNA, and 24-nt siRNA precursors
By mapping small RNA present in databases (Rajagopalan et al. 2006) to the complete collection of 76 npcRNAs (Supplemental Table 1; Hirsch et al. 2006), we revealed that 34 npcRNA loci give rise to at least one small RNA (Table 1 and Supplemental Fig. 2, npcRNA spanning small RNA loci). Some were already characterized small RNA precursors, such as the tasiRNA precursor TAS3 (npc41) or the MIR162A precursor (npc78) (Hirsch et al. 2006). The npc83 and npc521 corresponded to the miRNA precursors MIR869A and MIR160C, respectively. In addition, we identified new small RNA precursor candidates (npc34, npc351, npc375, npc520, and npc523). The majority of siRNAs deriving from these five npcRNAs are 24 nt long and map to both DNA strands of the npcRNA region, suggesting that these npcRNAs correspond to 24-nt-small RNA precursors. Expression analysis in different organs (Fig. 1) revealed that npc351 is enriched in stems and npc34 in aerial parts, whereas npc375 and npc523 show maximal levels in flowers. Specific oligonucleotides for real-time PCR studies could not be obtained for npc520. Whereas the npc375 and npc523 levels appeared unchanged in the rdr2 rdr6 double mutant, the npc34 and npc351 were up-regulated in both seedlings and flowers of rdr2 rdr6 (Fig. 1, right panel), suggesting that RDR2 or RDR6 produces a dsRNA from these npcRNAs, consistent with small RNA production from both DNA strands of these loci.
The Arabidopsis npcRNA collection and their matching small RNAs
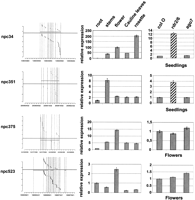
Expression patterns of four npcRNAs corresponding to new 24-nt siRNA precursors. (Left panels) The gray boxed regions (above or below are Watson and Crick strands, respectively) indicate the npcRNA gene and transcript (black line) on the Arabidopsis genomic DNA coordinates. Small RNAs identified in public databases are indicated by small bands on both strands. The size of each siRNA is indicated in Supplemental Figure 2; npcRNAs spanning small RNA loci (following a color code). (Middle panels) Expression of these npcRNAs in different tissues (roots, stems, flowers, cauline leaves, and rosette leaves) determined by qRT-PCR. Relative expression levels were normalized with ACT2 (AT3G18780), and values for roots were arbitrarily fixed to 1; SDs are shown. (Right panels) Expression in flowers or seedlings of Col-0 and a rdr2/6 or ago7 mutant. Values for Col-0 were fixed to 1.
Transcriptomic analysis identifies a novel DCL4-processed young miRNA gene and a novel npcRNA overaccumulating in dcl1 mutants
In order to characterize further this set of npcRNAs, we constructed a “dedicated noncoding RNA biology” microarray (the “RIBOCHIP”) containing 274 different oligonucleotide probes (50–70 nt in length) corresponding to all 76 npcRNAs, the sense/antisense couples for those npcRNAs encoding antisense transcripts, and a large collection of npcRNA-related RNA-binding proteins (for a complete list of genes, their identification, and spotted oligonucleotides with their main characteristics, see Supplemental Table 2). At first, the RIBOCHIP was hybridized with RNA from wild-type and dcl1-9 inflorescences (Fig. 2). Among seven known miRNA precursors spotted on the RIBOCHIP, six (MIR160C, MIR162A, MIR164B, MIR166A, MIR166B, and MIR168A) were up-regulated in dcl1 inflorescences, consistent with DCL1-processed conserved miRNAs. In contrast, the nonconserved MIR869A precursor (npc83) did not overaccumulate in dcl1. This miRNA precursor has an unusually stable secondary structure (Z-score = 16.0) (Hirsch et al. 2006) and other small RNAs are produced from the long stem-loop of this precursor (Fig. 3). RT-PCR analysis performed on other dcl mutants revealed that the MIR869A precursor overaccumulates in dcl4 (Fig. 3C). These results strongly suggest that, like MIR822 and MIR839, MIR869A is a young miRNA gene and that its transcript is processed by DCL4 because it adopts a secondary structure closer to that of perfect dsRNA than that of a classic miRNA precursor processed by DCL1 (Rajagopalan et al. 2006).

Regulation of genes in the RIBOCHIP. (A) Total number of genes that were either induced (red bars) or repressed (green bars) in each analyzed condition. Conditions include expression in roots versus leaves (roots), roots of phosphate starved plants (−P), plants under water stress (−H2O), roots of plants treated with 150 mM NaCl (NaCl), and inflorescences of dcl1-9 mutants (dcl1). (B) Heat map showing expression level of each individual gene summarized in A. (*) npcRNAs identified by Hirsch et al. (2006); (**) npcRNAs identified in this study. Genes without asterisks correspond to additional regulated genes from the RIBOCHIP (annotation is indicated: coding sense counterparts of npcRNAs, RNA-binding proteins, miRNA precursors, and control genes).

The npc83 is the miR869A precursor and is up-regulated in dcl4 mutants. The npc83 transcript encodes several small RNAs, notably miR869 (A), that are produced from a highly stable RNA-secondary stem structure (Z-score = 16). The position of the miRNA and detected siRNAs on the stem-loop of this npcRNA is indicated in B. (C) Expression of the MIR869A precursor was analyzed in dcl1-9, dcl2, dcl3, and dcl4 mutants and Col-0 seedlings by real-time RT-PCR. Data was normalized with ACT2 (AT3G18780), and values for Col-0 were arbitrarily fixed to 1. Two biological replicates gave similar results, and a representative example (SDs of technical replicates) is shown.
In addition to npcRNAs corresponding to conserved miRNAs, the npc531, which was not previously linked to RNA silencing mechanisms or known to encode a miRNA, also was up-regulated in a dcl1-9 mutant, a difference confirmed using quantitative RT-PCR (Fig. 4A, left panel). No known miRNAs map to this gene, suggesting that, alternatively, it could be the target of a miRNA. We identified a putative miR319 target site in npc531 (Fig. 4A, right panel), but 5′ RACE-PCR analyses did not reveal any specific cleavage of this transcript in this site (data not shown).
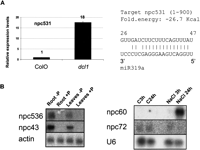
Expression analysis of npcRNAs. (A) Accumulation of npc531 transcripts in dcl1 mutants. Real-time RT-PCR expression analysis of npc531 in wild-type and dcl1-9 inflorescences (A) was normalized with the ACT2 (AT3G18780) gene, and values for wild type were arbitrarily fixed to 1. On the right, predicted pairing between npc531 and miR319a. (B) Regulation of npc43, npc60, npc72, and npc536 expression. Northern analysis was performed on the indicated npcRNAs under different stress conditions (plants grown in phosphate starvation or under salt stress).
Several npcRNAs are regulated by abiotic stresses
A deeper analysis of npcRNA accumulation was performed by hybridizing the RIBOCHIP array with RNA from wild-type leaves and roots, as well as roots of plants grown under stress conditions (phosphate starvation, salt stress, water stress) versus nonstressed control plants. Of the 274 genes spotted on the RIBOCHIP, which include the 76 npcRNAs (Supplemental Table 1; Hirsch et al. 2006), 42 were differentially expressed in at least one of the conditions assayed. From these 42, 26 corresponded to npcRNAs and only six to putative or known RNA binding proteins (RBPs) (Table 2; Fig. 2). The remaining regulated genes corresponded to cis-antisense coding transcripts or positive controls. The observed bias toward npcRNAs (62% of the regulated genes were npcRNAs) points to a dynamic regulation of these functional RNAs over npcRNA-related protein-coding genes, which can be additionally controlled at translational and post-translational levels. Moreover, several of these genes were regulated in more than one condition suggesting pleiotropic roles (Table 2).
Analysis of gene regulation using the RIBOCHIP
Independent validations of the results observed in the microarray experiments were obtained for several npcRNAs using Northern analysis (Fig. 4B; that also confirmed the predicted size for npc43, npc72, npc60, and npc536) and quantitative RT-PCR (Fig. 5). For the phosphate starvation assays, the up-regulation of npc43 and npc536 and the down-regulation of npc33 were confirmed (Fig. 5A). We also observed a slight up- and down-regulation of npc60 and npc311, respectively, which also were detected in the microarray experiments (albeit with low statistical significance). Salt stress resulted in a dramatic 100-fold increase in npc60 accumulation. For npc60, npc82, and npc536, the change in expression observed after 3 h of NaCl treatment was maintained after 24 h (Fig. 5B), whereas for npc72 the induction was transient (Fig. 5B). Differential expression of TAS3, npc43, and npc311 in roots and of npc15 and npc156 in leaves was confirmed, as we previously reported using semi-quantitative RT-PCR (Hirsch et al. 2006). Additionally, the MIR160C precursor was specifically enriched in roots, as already shown (Wang et al. 2005a). Taken together, the combination of microarray and quantitative RT-PCR allowed us to identify eight npcRNAs differentially expressed between roots and leaves and 15 npcRNAs regulated by diverse abiotic stresses.
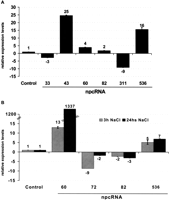
Regulation of specific npcRNAs in different stress conditions. Real-time RT-PCR expression analysis of indicated npcRNAs in roots of plants grown in phosphate starvation (A) and after 3h and 24h in 150 mM NaCl (B). In every case data was normalized with ACT2 (AT3G18780) and values for non-treated or wild-type controls were arbitrary fixed to 1. For each cDNA synthesis, quantifications were made in triplicate and two biological replicates were analyzed. Values are means ± SD.
Overexpression of two npcRNAs affects growth and differentiation in Arabidopsis
To explore the biological function of npcRNAs, we selected 12 candidates for further functional analyses. These candidates were chosen considering several criteria, which included conservation at the nucleotide level independent of encoded sORFs, the presence of statistically significant RNA secondary structures, tissue-specific expression patterns, and stress-regulation (Table 3). As a first step to elucidate the function of these 12 npcRNAs, we overexpressed the complete cDNAs under the control of the strong constitutive cauliflower mosaic virus 35S promoter in Arabidopsis. Overexpression of 11 out of these 12 npcRNAs did not lead to obvious visible differences compared with wild-type plants in nonstressed growth conditions (for a list of the overexpressed npcRNAs and the number of independent lines analyzed, see Table 3). In contrast, T1 plants overexpressing the npc48 showed drastic developmental anomalies, including an increase in the rosette diameter, leaf serration, and a delay in the flowering time compared with wild-type plants (Fig. 6A). Quantitative RT-PCR analysis of npc48 accumulation revealed that all transgenic lines exhibiting this phenotype overaccumulated npc48 (Supplemental Fig. 3). The phenotype of 35S∷npc48 plants resembled that of 35S∷MIR168 plants, which have reduced levels of the miR168 AGO1 target, and of AGO1-sensitive miRNAs such as miR166 (Vaucheret et al. 2006). 35S∷npc48 plants also showed a decrease in miR166 accumulation, but accumulated AGO1 mRNA and miR168 at wild-type levels (Fig. 6B). In addition, 35S∷npc48 plants, but not 35S∷MIR168 plants, exhibited reduced accumulation of miR164 (Fig. 6B), suggesting that npc48 could regulate a subset of miRNAs. T-DNA insertions within npc48 are not available.
Characteristics of selected candidate genes for functional analysis
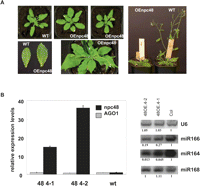
Phenotypic and molecular analysis of transgenic plants overexpressing npc48. (A) Phenotype of Arabidopsis plants transformed with 35S∷npc48 construct. A wild-type (WT) plant (left) and two representative T1 35S∷npc48 plants (OEnpc48) from independent lines displaying a characteristic serrated leaf phenotype at different growth stages (central photos) are shown. Rosette leaves from WT and OEnpc 48 plants (lower left panel). Image of whole plants (right panel) illustrates the delayed flowering of OEnpc48 plants. (B) Expression analysis of npc48 and AGO1 mRNA from leaves of two npc48 overexpressing lines and control WT using real-time RT-PCR (left panel). Northern blot analyses of miR164, miR166, miR168, and U6 RNAs (right panel). Quantification is indicated setting the Col-0 value arbitrarily to 1 and normalizing to U6 values.
As the expression of several npcRNAs was affected by environmental stress, we explored the response to salt stress and phosphate starvation in lines overexpressing npc43, npc60, npc311, and npc536. 35S∷npc536 transformants that actually overaccumulate npc536 (Supplemental Fig. 4) displayed heightened root growth under salt stress conditions (Fig. 7) compared with wild-type plants. This increase is due to both primary root growth and secondary root length under salt stress (100 and 125 mM), whereas no differences were observed in the absence of salt treatment. No phenotype could be observed in mutants that carry T-DNA insertions within npc536 (data not shown). The npc536 transcript is antisense to the AT1G67930 mRNA (encoding a Golgi-transport complex related protein) (Fig. 7C). The npc536 has a large dynamic range of expression across a wide range of tissue and hormonal, biotic, or abiotic treatment, whereas AT1G67930 is much less variable (AtGenExpress atlas) (Schmid et al. 2005). In response to certain abiotic stresses such as drought or cold treatment, the root expression of these two genes tends to be anti-correlated (Pearson correlation coefficient of −0.636 and −0.983, respectively). However, accumulation of the AT1G67930 transcript was not significantly modified in 35S∷npc536 plants or in npc536 mutants (Supplemental Fig.4). It remains possible that npc536 regulates the translation of AT1G67930 mRNA. Altogether, overexpression of two of 12npcRNAs affected Arabidopsis differentiation and growth responses to abiotic stresses.

Phenotypic and molecular analysis of transgenic plants overexpressing npcRNA536. Phenotype of Arabidopsis plants transformed with 35S∷npc536 constructs (Oenpc536). (A) Control and a representative OEnpc536 transgenic plants grown under the indicated salt stress conditions (0 mM, 100 mM, and 125 mM). (B) Quantification of primary root length and lateral root/primary root lengths in two independent transgenic OEnpc536 and control lines grown in 100 mM salt (n < 45 per experiment). SDs are indicated. These differences are statistically significant (t-test, 7 × 10−7), whereas no differences could be detected under normal growth conditions. (C) Schematic of the genomic positions and transcripts deriving from the npc536 (AT1G67920, white box) and AT1G67930 (dark gray box) loci. The nucleotide sequence of the overlapping region (light gray box) between the two transcripts is indicated.
Discussion
Through this work we revealed a variety of Arabidopsis non-protein-coding transcripts, some of which likely regulate development and responses to abiotic stresses. Whole-genome mapping in the model plant Arabidopsis, based on the use of tiling arrays, revealed that >50% of observed transcription was intergenic and that numerous antisense RNA transcripts exist (Yamada et al. 2003). In Drosophila, related experiments based on tiling-arrays, performed at six developmental stages, detected RNA expression for 41% of the probes in intronic and intergenic regions (Stolc et al. 2004). In agreement, analysis of EST and full-length cDNA libraries identified many transcripts not previously assigned to genomic loci (Okazaki et al. 2002; Numata et al. 2003; Ota et al. 2004; Riano-Pachon et al. 2005; Hirsch et al. 2006). At least 13% and 26% of the unique full-length cDNAs in mice and humans, respectively, are thought to be poly(A) tail–containing mRNA-like npcRNAs. Since the functions of most of these npcRNAs are unknown, much work needs to be done to identify regulatory RNAs among these genes.
Using genome-wide mapping of full-length cDNAs, we describe 33 new npcRNAs that, when added to the 43 previously identified (Hirsch et al. 2006), make a set of 76 npcRNAs dispersed throughout the Arabidopsis genome. These npcRNAs belong to at least five classes:
-
Three npcRNAs correspond to miRNA precursors such as the MIR869A precursor that overaccumulates in dcl4 mutants, suggesting that MIR869A is a young miRNA gene, similar to MIR822 and MIR839 (Rajagopalan et al. 2006).
-
Five npcRNAs give rise to 24-nt siRNAs that map to both DNA strands, suggesting that these npcRNAs define 24-siRNA precursors. Among these five npcRNAs, two (npc34 and npc351) overaccumulate in rdr2/6 mutants, suggesting that these npcRNAs are likely RDR substrates or, alternatively, targets of RDR-dependent small RNAs such as the tasiRNAs.
-
Five npcRNAs correspond to small RNA classes other than 21, 22, or 24 nt reminiscent of the class of 30- to 40-nt long-siRNAs (lsiRNAs) (Katiyar-Agarwal et al. 2007) recently shown to be involved in gene regulation in Arabidopsis.
-
Fourteen npcRNAs corresponded to cis natural antisense transcripts (cis-NATs), based on partial complementarity to other endogenous RNAs. The cis-NATs transcripts are derived from the same genomic loci as their sense counterparts, but from opposite strands. Genome-wide computational and experimental studies have shown that ∼5%–10% of gene transcripts in mammals and plants have cis-NATs (Wang et al. 2005b; Henz et al. 2007). cDNA sequence cluster analyses revealed that 7600 annotated genes in Arabidopsis (30%) had significant antisense expression (Yamada et al. 2003; Chekanova et al. 2007). Furthermore, characterization of 32,000 full-length rice cDNAs identified 600 antisense transcript pairs, half of which have no ORF in one member of the pair (Osato et al. 2003). Analysis of their regulation in comparison with their complementary transcripts indicates that there is a trend toward anticorrelated expression of cis-NAT pairs in Arabidopsis. However, currently available data do not produce a strong signature of small RNA mediated silencing for this process (Henz et al. 2007). The Arabidopsis transcriptome also contains a fairly large number of trans-NATs, ∼1320 putative trans-NAT pairs. Among those with available expression data, more than 85% were found in the same tissue as their sense partners (Wang et al. 2006). Several npcRNAs pairs of sense-antisense are coregulated, and potential NAT-based regulation systems in plants could be especially relevant for genes involved in developmental control or adaptive responses to changing environmental conditions. This assumption has recently been supported by the detection of NAT-specific small RNAs in plants that appear when antisense transcription is induced by salt stress (Borsani et al. 2005) or following pathogen attack (Katiyar-Agarwal et al. 2006). The npc536 forms a cis-NAT with AT1G67930. Overexpression of npc536 allowed plants to grow under salt stress but did not modify AT1G67930 mRNA accumulation. Furthermore, T-DNA mutants in this gene do not show misregulation of the antisense transcript. As this gene contains a sORF conserved in rice, it may act through this encoded peptide. Alternatively, it is possible that npc536 regulates translation of AT1G67930 mRNA or that it acts as a trans-NAT with a yet to be identified gene that plays a role in salt stress.
-
The remaining npcRNAs did not correspond to known classes of small RNAs and were not antisense to known protein-coding RNAs. One possibility is that they act through the production of short peptides. Computational approaches based on an analysis of codon bias and cross-species conservation suggest that ∼5% of annotated genes in the genomes of yeasts, plants, flies, nematodes, mice, and humans contain sORFs that could, in fact, be translated into exceptionally small peptides (Kastenmayer et al. 2006). Recently Kondo et al. (2007) reported that polished rice (pri), which was identified previously as a gene for a noncoding RNA in Drosophila, is in fact transcribed into a polycistronic mRNA that contains evolutionarily conserved sORFs that encode 11- or 32-amino-acid-long peptides. The small PRI peptides seem to act noncell autonomously to promote changes in epithelial-cell morphology. Hence, some Arabidopsis npcRNAs could encode sORFs that have regulatory roles. Alternatively, the RNA molecule may integrate ribonucleoprotein particles (RNPs) to determine the functional specificity of the complex (Prasanth and Spector 2007; Filipowicz et al. 2008) and orchestrate activities of other protein subunits during development (Brosius 2003; Wang et al. 2008). Other npcRNAs, such as meiRNA in yeast (Saccharomyces cerevisiae) and ENOD40 in legume plants, are required for correct subcellular localization of RNP particles (Yamashita et al. 1998; Campalans et al. 2004). Finally, npcRNAs could also play roles in transcription-dependent mechanisms rather than being RNA-sequence dependent per se (e.g., genomic imprinting, intergenic transcripts in the Drosophila bithorax complex; Prasanth and Spector 2007).
To explore the role of npcRNAs in Arabidopsis metabolism and physiology, we developed an array tool, the RIBOCHIP. This dedicated microarray contains 274 probes, representing not only the 76 identified npcRNAs but also coding transcripts for many npcRNA-related proteins such as RNA-binding protein genes, RNA-metabolism genes, small RNA biogenesis pathways genes, and other genes encoding proteins potentially linked to npcRNAs. Our RIBOCHIP analyses revealed a bias toward the regulation of npcRNAs over npcRNA-related protein-coding RNAs during stress conditions. One explanation for this bias is that npcRNAs, unlike protein coding RNAs, cannot be subjected to additional translational or post-translational controls that follow transcriptional regulations. Expression of many npcRNA candidates was regulated by growth conditions affecting root tissues, as seen for npcRNAs of the AtIPS1/At4 family (Franco-Zorrilla et al. 2002), and the ENOD40 family in legumes (Campalans et al. 2004). We propose that npcRNAs are candidate regulators to adapt root growth and development to soil biotic and abiotic interactions.
Additional analysis was done by overexpression of 12 npcRNAs in Arabidopsis. The overexpression of each npcRNA was verified in order to discard any silencing effect due to transgene expression. Overexpression of npc48 led to leaf serration, a phenotype that has been observed in several Arabidopsis mutants such as se or ago1. SE is a general regulator of miRNA levels, affecting the processing of primary miRNA precursors to miRNAs (Lobbes et al. 2006), while AGO1 acts as the major miRNA target slicing enzyme. Despite the resemblance of 35S∷npc48 plants with hypomorphic se and ago1 mutants, overexpression of npc48 did not have a general effect on miRNAs or AGO1 accumulation. However, miR164 accumulation was significantly impaired. Interestingly, mir164a mutants display abnormal serrations of the leaf margins (Nikovics et al. 2006). Minor differences in global miR164 accumulation were observed in mir164a mutants, and localized misregulation of miR164 targets appeared critical for this phenotype. Nevertheless, the phenotype of mir164a mutants is milder than the strong leaf serrations induced by overexpression of npc48, suggesting that the reduction in miR164 alone cannot account for the phenotype. Highly serrated leaves are a symptom of defects in cell proliferation along the margins of leaf primordia (March-Diaz et al. 2007), and the npc48 might be linked to this process. Recently, it has been shown that the npcRNA At4 inhibits in trans the action of miR399 through target mimicry (Franco-Zorrilla et al. 2007) without affecting miR399 accumulation. No homology or complementarity was detected between npc48 and any miRNA to suggest this. One possibility could be that npc48 partially inhibits post-transcriptional regulation by miRNAs using another mechanism.
Inhibition of lateral root growth has been suggested to be an adaptive response to environmental stresses, notably drought and salt stress (Deak and Malamy 2005; Xiong et al. 2006). The hormone ABA has been implicated in this response, although ABA independent pathways also exist. In addition, the cell layers surrounding the lateral root primordia conditioned the emergence of the lateral root, a process regulated by auxin signaling (Swarup et al. 2008). The overexpression of npc536 could modify the expression of specific target genes in this cell layer or affect ABA responses in lateral roots in order to modulate the length of the lateral roots under a particular stress condition. This gene is also induced by phosphate starvation, another condition affecting root architecture, and may play a role in the adaptation of root growth and development to the soil environment.
Although little is known about the biochemical activities of npcRNAs, an emerging hypothesis is that npcRNAs (long or small) incorporate into RNPs to determine their function and/or localization. Additional analyses are necessary to reveal the molecular mechanisms that underlie the action of npcRNAs in eukaryotes. Our work offers new perspectives on the action of npcRNAs, which are a sensitive component of the transcriptome, and reveals alternative riboregulatory mechanisms likely employed during plant growth and differentiation.
Methods
Plant growth and RNA extraction
All experiments used the Columbia (Col-0) ecotype of A. thaliana. Seeds of the silencing-related mutants—ago7-1, dcl1-9, rdr2/6, dcl4-2, dcl2-1, and dcl3-1—have been described before (Gasciolli et al. 2005; Adenot et al. 2006; Bouche et al. 2006; Vaucheret 2006). Plants were grown in long day conditions (16-h light/8-h dark photoperiod) with 150 μmol m−2 sec−1 of supplemental fluorescent light at 23°C.
For analysis of gene expression under salt stress, plants were grown in hydroponics in liquid 0.5× Murashige and Skoog (MS) salts (Sigma), 1% sucrose, during 2 wk (until roots were 5–8 cm long). Then, the medium was replaced by fresh 0.5× MS (control) or 0.5× MS containing 150 mM NaCl. After 3 h and 24 h of incubation, roots of ∼100–120 plants were separated from aerial parts and pooled for RNA extraction. Phosphate starvation treatments were performed according to the method of Franco-Zorrilla et al. (2002). Fourteen days after germination, roots of at least 100 plants were pooled and total RNA extracted. Water stress conditions were performed as described by Manavella et al. (2006). For analyzing stress responses, 45–50 plants of T1 segregating lines overexpressing an npcRNA were sterilized and germinated for 2 d and then transferred to a medium containing NaCl or not as indicated. After 14 d, primary and lateral root length for each plant are individually measured and photographed. All seedlings are transferred to the greenhouse and Basta selection and PCR-mediated genotyping determined homozygotes, heterozygotes, and wild-type plants from the lot. Experiments were done per triplicate.
Inflorescences, stems, and leaves of wild-type and mutant plants were collected from greenhouse-grown plants. Seedlings were grown in vitro for 3 wk. Total RNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s instructions and further purified by passage through RNAeasy columns (Qiagen) and residual genomic DNA removed by on-column DNase I treatment (Qiagen).
Bioinformatic analysis of npcRNAs
To identify candidate npcRNAs from the “full length” sequenced cDNA databank (Castelli et al. 2004), RNAs were first mapped with Sim4 (Florea et al. 1998) on the genome and selected by the length of the longest ORF (<210 nt). About 500 candidates were further tested for lack of significant BLAST hits against NCBI nr protein databank. Mapping of other ESTs and cDNAs on these genomic regions did not bridge the gap with already annotated neighboring genes. Mapping of short RNAs (sRNAs) on the total set of 76 npcRNA was performed using a large data set of massively sequenced A. thaliana sRNAs (Rajagopalan et al. 2006). Potential miRNAs regulating npc531 were searched in miRBASE database. Sequence comparisons were performed using BLASTN for nucleotide sequences and BLASTX for encoded sORFs sur TAIR, NCBI nonredundant and chromosome databases. Similarities between nucleotide (BLASTN) and sORF-encoded peptide (BLASTX) were searched up to an expected value E = 1 × 10−5. Boxes of at least 21 nt conserved between number of species N independent of the encoded ORFs are indicated as N(+). Conserved sORFs are indicated as N(XX), where N is the number of species and XX is the size of the sORF in A. thaliana. Those cases where the sORF show similarity with a larger ORF (>100 amino acids) in other species are indicated as putative pseudogenes (pp). Exceptionally stable RNA secondary structures were predicted as by Hirsch et al. (2006). The sequences were scanned using sliding windows of increasing lengths. Briefly, for each window, the free-energy of folding of the most stable structure was computed using rnaFold from the Vienna Package (http://www.tbi.univie.ac.at/~ivo/RNA/) and compared with the distribution of free-energy of folding of random sequences similar in size and nucleotide composition. The Z-score, defined as the difference between these free-energies divided by the standard deviation, was calculated, and sequence regions with a Z-score > 5 were displayed in Supplemental Figure 1.
RIBOCHIP array design
Oligonucleotide primers were designed using OligoWiz 2.0 (Wernersson and Nielsen 2005). Each 45–60mer oligonucleotide probe was printed as triplicates on Quantifoil QMT epoxy slides in 16 grids in a 4 metacolumns × 4 metarows configuration with a 9 columns × 7 rows spot pattern for each subarray by the Institute of Genome Research, Center of Biotechnology at Bielefeld University, Germany. Additionally, the array contains probes specifically designed as labeling and hybridization controls (positive) corresponding to nine A. thaliana genes: AT5G44200 (nuclear cap-binding protein), AT1G07920 (EF1-alpha), AT5G03240 (UBQ3), AT4G05320 (UBQ10), AT3G13920 (EIF4A1), AT1G19910 (V-type H+ ATPase 16 kD subunit), M21415 (TUB4), AT1G49240 (ACTIN 8 protein), and AT3G18780 (ACTIN 2 protein). At least two independent biological replicates were performed for each condition analyzed in the RIBOCHIP.
Target preparation for RIBOCHIP array
Two micrograms of total RNA from each biological sample (treatments and controls) were amplified using the Amino Allyl Message Amp II aRNA Amplification Kit (Ambion), following the protocol provided by the manufacturer. Ten micrograms of amplified RNA (aRNA) was chemically labeled with Cy5 (Q15108, Amersham), monofunctional reactive dye or Cy3 (Q13108, Amersham), monofunctional reactive dye in coupling buffer (Ambion). Labeled aRNA was purified from noncoupled dyes using the columns supplied by the Amino Allyl Message Amp II aRNA Amplification Kit.
Array hybridization
Before use, the printed slides were processed according to the manufacturer’s recommendations (Institute of Genome Research, Center of Biotechnology, Bielefeld University) and then hybridized with 200 pmol each of Cy5- and Cy3-labeled targets (treatment and control) in 5× SSC (0.75 M NaCl, 75 mM sodium citrate at pH 7), 25% formamide, 0.1% SDS for 60 h at 42°C. For each slide, 20 μg of salmon sperm DNA and 4 μg of yeast tRNA (Sigma) were added as blocking agents. After hybridization slides were washed once at 42°C in 1× SSC, 0.2% SDS for 4 min followed by a wash at room temperature in 1× SSC, 0.2% SDS for 4 min and twice at 4 min each in 0.1× SSC. A quick final wash at room temperature in 0.05× SSC for 1 min was done to remove any salt residue and particles bound to the slides.
Slides were processed using an Axon 4000B microarray scanner (Axon Instruments) with 635-nm and 532-nm lasers and a scan resolution of 10 μm to generate TIFF images on each channel. PMTs for lasers were automatically set by software for each slide (GenePix 6.0, Axon Instruments).
Microarray data processing and normalization
Quantification of images on 635-nm and 532-nm channels was performed with GenePix 6.0 software (Axon Instruments). Normalization was performed by the Loess and median by block method using MANGO software developed at the Gif-Orsay DNA microarray platform (Center de Génétique Moléculaire, CNRS). Replicate spots outliers were detected by Grubb’s test. Statistical significance of regulated probes was analyzed by the single slide methods of Newton et al. (2001), Sapir and Churchill (2000), and Chen et al. (1997).
Gene expression analysis
For real-time RT-PCR, cDNA was synthesized by reverse transcription performed on 1.5 μg of total RNA using the SuperScript II Reverse Transcriptase (Invitrogen) and (T)16 A/G/C oligonucleotides. Real-time RT-PCR was performed on the Roche Light Cycler instrument using SYBR Green I dye (LightCycler FastStart DNA MasterPLUS SYBR Green I, Roche) using the primer pairs listed in Supplemental Table 3. Technical triplicates were done for each assay, and at least two independent biological replicates were assayed.
5′ RACE-PCR assays to detect npcRNA 531 cleavage products and Northern analysis of small RNAs were performed as described by Hirsch et al. (2006).
Construction of A. thaliana npcRNA overexpression lines
The coding sequences for each npcRNA were amplified by PCR from genomic A. thaliana Col-0 DNA using the primers showed in Supplemental Table 4. The full-length DNAs were cloned into pENTR/D-TOPO (Invitrogen) and then in the pB7WG2 binary plasmid (Karimi et al. 2002), under the control of CaMV 35S promoter, using the gateway technology (Invitrogen) to be introduced into Agrobacterium tumefaciens (agl-o). These constructions were used to transform A. thaliana ecotype “Columbia” plants, by floral dip (Bechtold and Pelletier 1998). Transgenic plants were selected by spraying seedlings from T1 generation 7, 9, and 11 d after germination with a solution of 0.01% Basta (200 g/L glufosinate ammonium). Basta-resistant plantlets (T1) were then grown in soil under long-day conditions (16-h light). Expression levels of the transgene in the T1 and T2 generations were determined by qRT-PCR.
Acknowledgments
We thank Raquel Chan (Argentina) for water stress RNA samples and Javier Paz Ares (Spain) for helpful discussions. P.L. is the recipient of a fellowship from the Ministère de l’Education Nationale et de la Recherche (MENR; France); S.W. was a fellow from the Fundacion Foro de Biotecnologia, Argentina. F.M. was supported by the Ministerio de Educacion y Ciencia, Spain. This work was also supported by the European FP6 RIBOREG project no. LSHG503 022 and partly by the French ANR-Genoplante RIBOROOT project.
Footnotes
-
↵7 Present addresses: Laboratorio de Agrobiotecnología, Piso 2, Pabellón 2, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Buenos Aires, Argentina;
-
↵8 INRA, Campus International de Baillarguet, UMR BGPI, TA A-54/K 34398, Montpellier, France;
-
↵9 Alten Ouest, 12i Rue du Patis Tatelin, ZAC Saint-Sulpice, 35000 Rennes, France.
-
↵10 Corresponding author.
↵E-mail crespi{at}isv.cnrs-gif.fr; fax 33-1-69-82-36-95.
-
[Supplemental material is available online at www.genome.org.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.080275.108.
-
- Received April 30, 2008.
- Accepted October 7, 2008.
- Copyright © 2009, Cold Spring Harbor Laboratory Press








